
More Information
Submitted: February 14, 2025 | Approved: February 21, 2025 | Published: February 24, 2025
How to cite this article: Das V, Bhatia M, Pradhan J. Critical Understanding of Apoptosis’ Function in Diabetes and Diabetic Wound Healing: Prospective Therapeutic Opportunities. J Stem Cell Ther Transplant. 2025; 9(2): 012-026. Available from:
https://dx.doi.org/10.29328/journal.jsctt.1001047
DOI: 10.29328/journal.jsctt.1001047
Copyright License: © 2025 Das V, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Apoptosis; Apoptosis markers; Diabetes; Diabetic wound; Treatment strategies
Abbreviations: T2DM: Type-2 Diabetes Mellitus; PS: Phosphatidylserine; FLMP: Formyl-methionyl-leucylphenylalanine; HETEs: Arachidonic acid derivatives having hydro-peroxy moieties; CAD: Caspase-activated DNAse; PT: Permeability Transition; AIF: Apoptosis-Inducing Factor; ICE: Interleukin-1 Converting Enzyme; PARP: Poly-Adenosine-Diphosphate-Ribose Polymerase; ErbB: Epidermal growth factor receptor family; AV: Autophagic vesicles; IFN: Interferon; ILs: Interleukins; MEF: Mouse Embryonic Fibroblasts; PI3K: Phosphotidyl-Inositol-3-Kinase; mTOR: Mammalian Target of Rapamycin; XIAP: X-linked Inhibitor of Apoptosis Protein; ARHI: Aplasia Ras Homolog Member-I; LPS: Lipopolysaccharide; NUCB2: Nucleobindin-2; PC: Prohormone/pro-protein convertase; SP: Signal Peptide; C’3b/C’5a: Complement Factors; CSFs: Colony Stimulating Factors; FXlI: Hageman Factor; FGF: Fibroblast Growth Factor; MCP-1: Monocyte Chemotactic Protein-I; PAF: Platelet-Activating Factor; PDGF: Platelet-Derived Growth Factor; PG1, 2, D, E, F: Prostaglandins; TNF: Tumor Necrosis Factor; LSC: Laser Scanning Cytometry; HAM: Human Amniotic Membrane; AECs: Amniotic Epithelial Cells; AMCs: Amniotic Mesenchymal Cells; HCV: Hepatitis C Virus; NOS: Nitric Oxide Synthase; ER: Endoplasmic Reticulum; AV: Autophagic Vesicles; FADD: Fas-Associated Death Domain; DN: Diabetic Neuropathy.

Critical Understanding of Apoptosis’ Function in Diabetes and Diabetic Wound Healing: Prospective Therapeutic Opportunities
Vishnu Das1, Mamta Bhatia2 and Joohee Pradhan1*
1Department of Pharmaceutical Sciences, Mohanlal Sukhadia University, Udaipur, Rajasthan 313001, India
2JIET College of Pharmacy, Jodhpur, Rajasthan, India
*Address for Correspondence: Joohee Pradhan, Department of Pharmaceutical Sciences, Mohanlal Sukhadia University, Udaipur, Rajasthan 313001, India, Email: [email protected]
Apoptosis, or programmable necrosis, can be induced by miscellaneous factors such as radiation, chemicals, and physiological and pathological conditions. Apoptosis is required for many processes, such as the turning over of normal cells, and the growth of the immune system and functions, unfair apoptosis either too little or too much has been linked to several clinical circumstances, accompanied by cancer, diabetes, and neurodegenerative spoiling. The harm of β-cell cytoplasm owing to pancreatic cells dying (T2DM) is a complicated etiology of Non-insulin-dependent diabetes mellitus (type II). To halt the normal progression of pancreatic cell disorders, it may be required to modulate the apoptosis and proliferation processes of these cells. Apoptosis is a complex process with four major components: induction, detection, effectors, and eradication. Each step necessitates the coordinated action of multiple molecules, the most noteworthy of which are caspases, the Bcl-2 protein family, and p53 (tumor suppressor gene).
With a greater understanding and a comprehensive search through the journal’s databases, and the molecular activities of these biochemical apoptotic markers, it may be feasible to create innovative techniques for the treatment of diabetes and its consequences.
This review presently explains the complications of insulin-dependent and non-insulin-dependent diabetes that may result in angiopathy complications such as diabetic neuropathy, diabetic encephalopathy, diabetic retinopathy, and diabetic wounds. New studies that defined some of the most significant activities of apoptosis in the treatment of wounds were reviewed.
This review highlights the importance of caries by considering the intricate molecular pathways involved in apoptosis, particularly the roles of caspases, Bcl-2 protein clan, p53 varying the care of diabetic wounds and diabetes, and modulation in the supervision of diabetes and diabetic wounds. This underscores the potential for future research to explore and harness the beneficial effects of modifying apoptosis for improved diabetes management strategies.
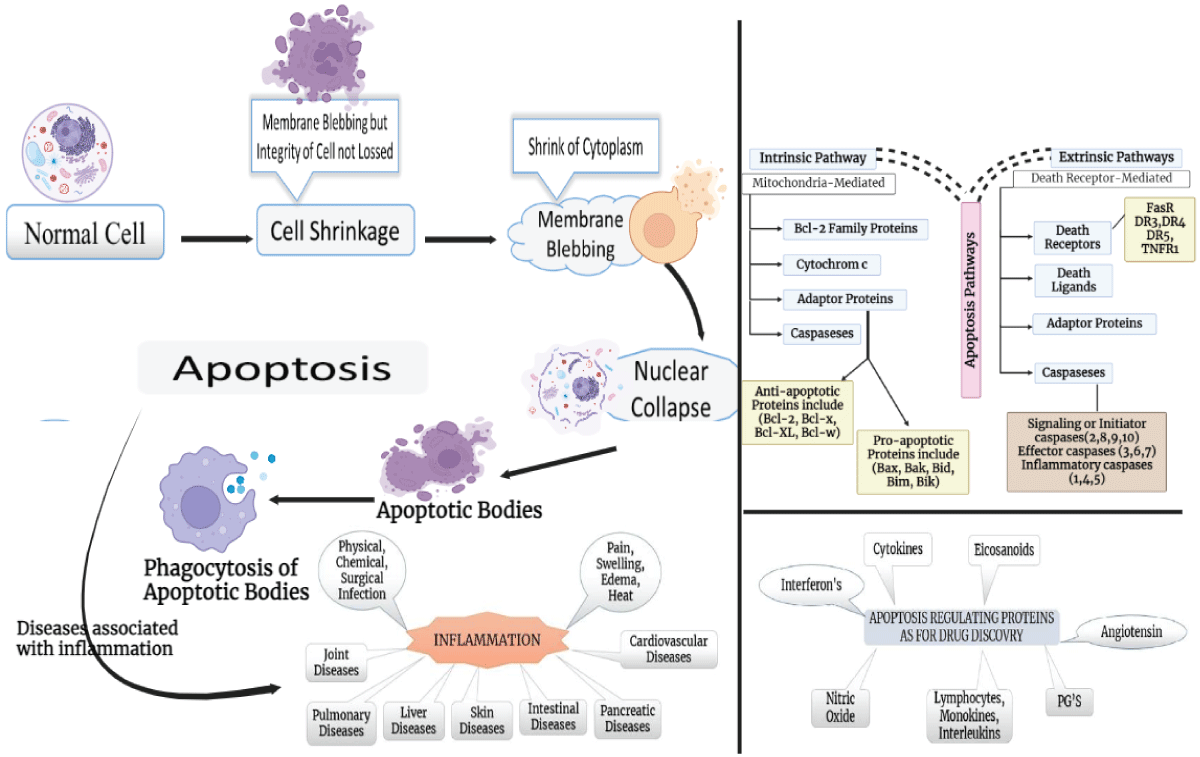
Graphical abstract.
A human adult loses 50-80 billion cells every day as a result of apoptosis. This biological mechanism successfully eliminates contaminated cells, precancerous cells, and other forms of cancer cells while preserving cell balance in the body. Cell death occurs naturally in the liver, but it has also been related to hepatic disorders such as bile salt-induced apoptosis. Alcohol causes apoptosis in both the liver and the pancreas, which has been related to pancreatic beta cell harm in insulin-dependent diabetes mellitus [1]. Although many different provocations and circumstances, both healthy and pathological, can cause cell suicide, not all cells will succumb and die from the common provocation.
Diabetes Mellitus (DM) is a habitual metabolic complaint characterized by patient hyperglycemia carried out from blights in insulin stashing, insulin action, or both. It is one of the most prevalent and debilitating diseases worldwide, affecting millions of people. The disease is classified into Type 1 Diabetes Mellitus (T1DM), an autoimmune disorder leading to β-cell destruction and insulin deficiency, and Type 2 Diabetes Mellitus (T2DM), which is characterized by insulin resistance and progressive pancreatic β-cell failure. Additionally, Gestational Diabetes Mellitus (GDM) occurs during pregnancy and may predispose both the mother and the child to the development of T2DM later in life [2]. Long-term hyperglycemia in diabetes results in various complications, including cardiovascular disease, nephropathy, neuropathy, and retinopathy, significantly impacting the quality of life and increasing mortality rates [3].
One of the most challenging complications of diabetes is impaired wound healing, which often leads to chronic ulcers and, in severe cases, limb amputation. Understanding the molecular mechanisms underlying these complications is crucial for developing effective therapeutic interventions [4].
Role of apoptosis in health and disease
Apoptosis, or programmed cell death, is a fundamental biological process that maintains tissue homeostasis by eliminating damaged, aged, or unwanted cells. This tightly regulated process is essential for normal development, immune function, and cellular turnover. It occurs through two primary pathways: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway, both of which culminate in the activation of caspases, a family of proteases responsible for orchestrating cell dismantling and removal [4].
While apoptosis is a necessary physiological process, its dysregulation is implicated in various pathological conditions. Excessive apoptosis leads to degenerative diseases, such as Alzheimer’s and Parkinson’s disease, while insufficient apoptosis contributes to uncontrolled cell proliferation in cancers. In diabetes, an imbalance in apoptosis significantly affects pancreatic β-cell survival, vascular integrity, immune function, and wound healing capacity, making it a crucial target for therapeutic intervention [5].
Importance of understanding apoptosis in diabetes and wound healing
In diabetes, excessive apoptosis contributes to progressive β-cell loss, leading to insulin insufficiency and metabolic dysregulation. Various factors, including oxidative stress, chronic inflammation, glucotoxicity, lipotoxicity, and Endoplasmic Reticulum (ER) stress, drive apoptosis in pancreatic β-cells. The destruction of these insulin-producing cells exacerbates hyperglycemia, accelerating disease progression [4].
Beyond β-cell apoptosis, diabetes is also associated with dysregulated apoptosis in endothelial cells, fibroblasts, keratinocytes, and immune cells, which significantly impacts wound healing. Diabetic wounds exhibit impaired angiogenesis, reduced Extracellular Matrix (ECM) deposition, and persistent inflammation, creating a hostile environment for tissue regeneration. This leads to the formation of chronic ulcers, increasing the risk of infections and amputations [2].
Therapeutic strategies aimed at modulating apoptosis hold great promise for preserving β-cell function, improving tissue regeneration, and enhancing wound healing in diabetic patients. Novel approaches, such as antioxidants, growth factors, gene therapy, and stem cell-based therapies, have demonstrated the potential to restore apoptotic balance. A deeper understanding of the molecular mechanisms underlying apoptosis in diabetes could pave the way for the development of innovative and targeted treatments to mitigate diabetes-related complications [3].
This review aims to explore the intricate relationship between apoptosis and diabetes, focusing on its impact on pancreatic β-cell survival and diabetic wound healing. Furthermore, it will highlight emerging therapeutic strategies that target apoptotic pathways to improve diabetes management and clinical outcomes [5].
Apoptosis and mechanisms
Mechanisms of cell death (apoptosis-I): Cell death receptors are used for extrinsic signaling. FLICE-inhibitory protein (FLIP) can impact susceptibility to FAS and TNF signaling competes with caspase 8 for FADD [6]. Despite several mechanisms indicating that FLIP carries regulatory action, its precise contribution to FAS Signaling remains unknown [7].
Mechanisms of cell death(apoptosis-II): Routes of intrinsic death are regulated under Bcl-2. Abnormally regulated cell death is allied with the pathophysiology of several spoiling [8]. The stress-induced route of cell caries is dependent on a sophisticated structure involving pro and anti-apoptotic proteins (Bcl-2 kin) that occurs prior to caspase activation, whereas the outer pathway leads straight to caspase activation. Proapoptotic Bcl-2 kin members (like Bax and Bak) recruit and oligomerize the outer mitochondrial membranes, whereas antiapoptotic Bcl-2 kin members simultaneously block the BH3 proteins, according to one commonly accepted explanation for the intrinsic pathway. Bax and Bak can form massive homo-oligomeric complexes in the mitochondria, which may act as a “pore” that releases Cytochrome-C. The CARD-containing Apaf-1 adaptor is subsequently activated by cytochrome-C, forming a heptameric scaffold for caspase 9 recruitment. The massive complex that develops, as a result, is known as the apoptosome, and it allows initiator caspases to be activated first, followed by effector caspases dimerization [9]. The perspective of the Bax/Bak-raised pores and indisputable importance of mitochondrial proximal function in stress-induced apoptosis are just two of the many aspects of this paradigm that are currently being debated. The complicated temporal-spatial management of three Bcl-2 families for the unification of squeeze and lysis-related signals of cell organ. All kin are covered as turned parts that follow. The effects of neem bark extract on cell death and pro-inflammatory cell signaling, aid in the help to explain the processes induced by Azadirachtaindica [10].
Mechanisms of cell death (apoptosis III): a prelude for the endoplasmic reticulum in cell suicide: The ER which is the primary storage region of intracellular calcium, is also involved in suitable protein folding. The folding of protein defects can cause “unfolded protein response” and cell lysis. Misfolded or mutant proteins that cause ER stress-induced apoptosis have been associated with Huntington’s and Alzheimer’s disorders. Recent studies have shown that Bcl-2 and Bax/Bak play opposing roles in ER stress-induced cell suicide. Bcl-2 kin blocks the mitochondria from receiving a stress signal from the ER. These findings highlight of complex precept of ER and mitochondria, with the contribution of calcium dynamics in cell lysis [11]. The two possible signaling modules discovered, one including the proteins CED-12/ELMO and CED-5/Dock180, if serve as bilateral GEF for Rac-1,
and the other involving CED-1/LRP1 [12]. Cell death starts with apoptosis and is followed by phagocyte engulfment as depicted in Figure 1. The increased output of inflammatory mediators and upregulation of numerous genes’ response to the recognition of the apoptotic bodies may cause phagocytes to become more capable of engulfing and/or eliminating the apoptotic-cell debris in sum to serving a correct “Filth disposal”. Deficits in the removal of death cells connected to the formation of an ailment state; for illustration, macrophages in atherosclerotic plaque have reduced the kinetics of death cell circumambiency. The elimination of death cells by the macrophages of Systemic Lupus Erythematosus (SLE) patients exhibits defects. Phagocyte deficits are also linked to autoimmune diseases. The engulfment process is the right way of potential therapeutic action because, according to common knowledge, issues with clearance lead to exposure to auto-antigens, which ultimately cause autoimmune disease. When removing corpses, several recognized stages are employed. A phagocytic cup arises when a phagocyte initially contacts an apoptotic cell; the target is explicitly identified, and continued signaling leads to action programmed and phagocytosis. The body of the death cell is finally destroyed once it has been completely absorbed. Ultra-structural examinations of engulfment reveal two unique mechanisms in which the phagocyte engulfs apoptotic cells. The anti-inflammatory condition that is distinct from that brought on by necrotic cells is established by changing the cytokine release of the phagocyte in response to the detection of the apoptotic cell [12]. Cells perish by programmed cell death in the growing embryo during morphogenesis and in the adult human/creature during or at the end of an immune action. Since apoptosis plays a crucial physiological role, its aberration may be harmful. Thus, an unscheduled program of cell death of some brain neurons contributes to diseases like Alzheimer’s and Parkinson’s while cancer results from proliferating cells’ inability to start apoptosis after suffering significant DNA damage [13]. The balance between cell death and growth must be maintained, which apoptosis does. This sophisticated process is carried out by two primary channels that collaborate to activate an executioner rule of procedure, which causes chromatin tearing and nuclear fragmentation. The balance between cell death and new cell development must be maintained, which apoptosis does. This sophisticated process is carried out by two primary channels that collaborate to activate an executioner process, which causes chromatin breakage and nuclear fragmentation. Deregulation of these routes is frequently implicated in the genesis of cancer and the progress of cancer treatment antagonism [14]. Apoptosis has recently been related to right ventricular dysplasia, QT syndrome, ischemic, idiopathic dilated cardiomyopathies, after infarction, and other cardiovascular conduction system anomalies [15].

Figure 1: Process of Apoptosis.
Programmed cell deaths are coordinated and typically utilize energy in a procedure that requires the activation of accumulation of cysteine proteases known as “caspases” with the puzzled cycle of phenomena linking the initial stimulation of the cell’s eventual death. Immune cells’ apoptotic activity is critical for inflammation decrease. The basic method by which cells are killed, apoptosis, has been related to the formation of diabetes mellitus via antigen cross-linked pathways culminating in the activation of specific T-cells. Caspase-3 is the principal caspase included in the apoptotic process [16].
Autophagy: Autophagic Vesicles (AV) encapsulate organelles and proteins, which are eventually destroyed by fusion with the lysosome. LC3 is a protein with Ubiquitin-like characteristics that attaches to AV membranes in cells during autophagy. Treatment with lapatinib with obatoclax resulted in the formation of major LC3 decided vesicles in HCT 116 cell transaction GFP-LC3.When compared to non-treated cells, the proportion of upbringing cells with major LC3-positive skeleton enlarges with single-agent treatments but enhances the addition of multi-agent therapies dramatically. In breast cancer cells, the addition of obatoclax and lapatinib was demonstrated to marginally boost just one isoform of LC3. The data on the siRNA trial provided the most persuasive evidence that autophagy is essential for cell death during treatment with a combination of targeted therapies. The autophagy proteins ATG5 and Beclin protect against cell death during treatment with lapatinib and obatoclax [17].
Macrophage- This pleiotropic cell type is favorable in the repair of normally healed inflammation/wounds, but it may induce excessive inflammation or fibrosis in other conditions. As per a certain study, the etiology of uncured and poorly healing wounds includes malfunctioning macrophages. Because of advances in our understanding of this versatile cell, the macrophage remains a promising therapeutic site, both to inhibit fibrosis and scarring and to improve chronic wound healing [18].
Apoptosis pathways
The cell type, stimulation, and other environmental factors play multiple roles in all apoptotic pathways. Apoptosis can be triggered by one of two mechanisms discussed in Figure 2, Table 1.

Figure 2: Apoptosis mechanism pathways (a) and (b): Apoptosis can be induced by mitochondria, which initiate the process by releasing cytochrome c into the cytoplasm.
| Table 1: Apoptosis pathways. | |
| Extrinsic Pathway | Intrinsic Pathway |
| This process is triggered by external signals like the attachment of ligands on death receptors present on the cell surface, headmost to apoptosis. The family of Tumor Necrosis Factor genes includes these receptors. Upon interaction with a receptor, caspases are activated. | In an intrinsic pathway, internal factors such as metabolic stress, DNA damage, and a lack of growth hormones, triggered this process. The controlling molecules for this pathway are Bax and Bcl-2. In response to stimulus, these chemicals determine whether a cell will live or perish. Bax and Bcl-2 are cytoplasmic proteins that promote and prevent apoptosis, respectively. |
Basic mechanism of apoptosis
When apoptosis is triggered, the mitochondrial protein stimulates caspase-9 activation by binding to IAPs. IBM-containing proteins’ IAP-neutralizing properties are expected to play a variety of activities, but are not delimited to, the precept of cell lysis. A plethora of IBM-Conveyance proteins suggests that accurate signaling selection beneath the physiological phase has yet to be established. In its entirety, IAPs are just signal modulators that may function as nodes in the integration and transformation of molecular acquaintance in the suitable biological currency of death and inflammation [19]. IAP antagonists are intriguing cancer therapeutic candidates since they can either directly kill cancer cells or augment current chemotherapy. Controlling the Bcl-2/Bax rheostat, which determines apoptosis susceptibility, is another potential technique for the therapy of cancer and other illnesses. Small compounds that imitate BH3 domains are already prototype pharmaceuticals, and their structure-activity relationship profile still has an opportunity for development. Furthermore, it is unclear whether Bcl-2 antagonists and BH3 mimics will have an effect all-encompassing on the various Bcl-2 family members [7]. Death receptors aid in the development of apoptosis by linking extrinsic. Caspase-8 and other effectors caspases are activated, and a death domain is recruited [9]. Activity-regulated cytoskeleton-associated protein (ARC) and FLIP suppress death-receptor-mediated cell death. In the unshed area of the picture, activator caspases such as caspase-3 occur where mitochondrial and death-receptor-mediated pathways overlap. Bcl-2-interacting protein that promotes cross-link in between the two pathways [20]. Cell proliferation-related genes like c-myc and p53 are among the many genes implicated in apoptosis regulation. Over time, it has clear that bcl-genes show an essential role in regulating apoptosis [21].
Caspases-8 crevice of the BH3-only protein Bid, for example, may become a crucial pathway in cells where there is insufficient active caspase-8 to allow direct crevice and activation of effector caspases. This Bid crevice originates the pro-apoptotic tBID bit, which triggers mitochondrial cytochrome-c liberation and caspase-9 exhortation. Caspases 9 and 8 can then bid each other, resulting in a positive loop that enhances initial caspase-8 signals. Two primary pathways direction apoptosis in human arrangement: extrinsic process started by the death gatherer and internal route driven by mitochondria (Figure 2). The two major routes of apoptosis induction are widely accepted to be extrinsic signaling by the death gatherer, which steerage to the building of the death propel signaling group (DISC), and intrinsic signaling primarily via mitochondria that steerage the fabrication in apoptosome. The creation of DISC and common effectors caspases may cause apoptosis. Both routes are active in the immune system; however, whether they are enough to maintain lymphocyte homeostasis is unknown. In lymphocytes, new apoptotic mechanisms, cum as caspase-unattached routes and granzyme-initiated pathways, recently have been found [22].
Understanding the processes that cause apoptosis and how it pertains to autoimmune ailments (Figure 3). T lymphocytes countenance several “life-or-death” checkpoints while they grow in the thymus, as naïve cells, and after maturation. Negative selection and the lack of selection cause around 95% of cells to die (left panel). To prevent cell death, naive T cells in the Circumference (right panel) desire cytokine support and T-cell receptor exhortation. T cells that are activated by pathogens proliferate before dying. Apoptosis is caused by the removal of growth factors, including intrinsic mechanisms and AICD (activation-induced cell death) via death receptors. Memory cells require cytokines to survive. When apoptosis fails at one of the critical checkpoints, two ailment phases can develop: autoimmunity or haematological malignancies. Identical checkpoints exist for B lymphocytes.
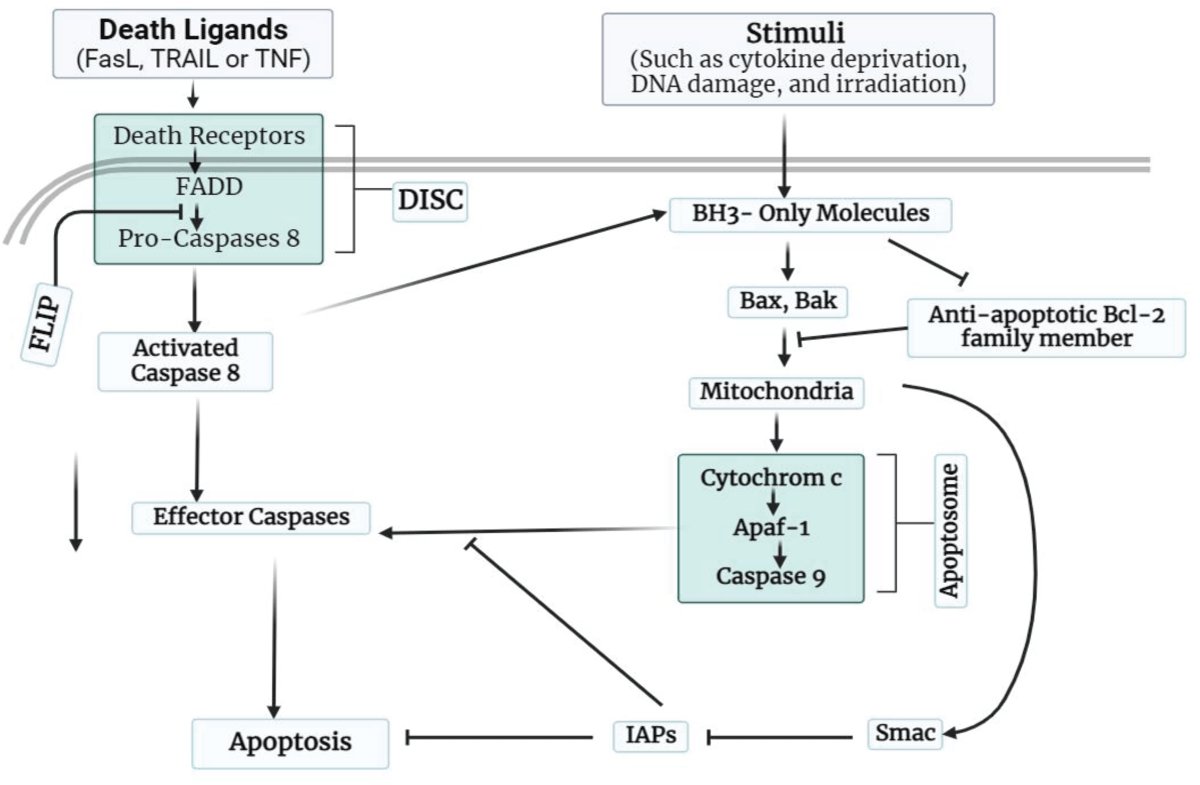
Figure 3: A schematic showing the intrinsic (left) and extrinsic (right) paths to apoptosis.
Figure 4 depicts the modeling of death gatherer extrinsic signaling by Fas-propel apoptosis. When Fas aggregates, death death-inducing signaling complex, which includes bi-functional FADD and activator caspase 8, is formed. Dimerization, activation, and high local caspase concentrations cause caspase 8 to be released from the membrane of the cell. The intrinsic signaling, route is shown on the right. In response to cell shrinkage signals, the pro-apoptotic Bax/Bak and BH3 proteins are translocated into mitochondria. Bcl-2 proteins counteract as defensive effects of Bcl-2 on cellular survival. As a result of Bax/accumulation Bak’s and the creation of large oligomers in the membrane of mitochondria, cytochrome c (Cyt c) is passed into the cellular fluid. When Cyt C is present, Apaf-1 forms the “apoptosome,” which provides significant scaffolding for caspase activation. Both processes activate effector caspases, which then cleave a variety of cellular substrates. Induction, detection, effector, and elimination are the four essential phases of the complex process known as apoptosis. During the prompting phase, the cell takes an apoptotic signal. Extrinsic causes for apoptosis include dietary habits, cytokine loss, radiation, and oxidative stress. Cell integrates a range of checkpoints acquired from signal transport route to adjudge, whether to make over apoptosis. After the cell decides to undergo death, a signal to activate the lysis mechanism is to be discovered and sent to downstream effectors. Some steps are followed to perform the apoptotic response, and finally cell is destroyed through phagocytosis. Caspases, the Bcl-2 proteins, and p53 are among many components that must collaborate to complete each stage of the apoptosis process [23]. Preliminary research indicates that type-2 diabetes and smouldering myeloma may also be efficiently treated with IL-1 targeting, expanding the breadth of IL-1-driven illnesses [24].
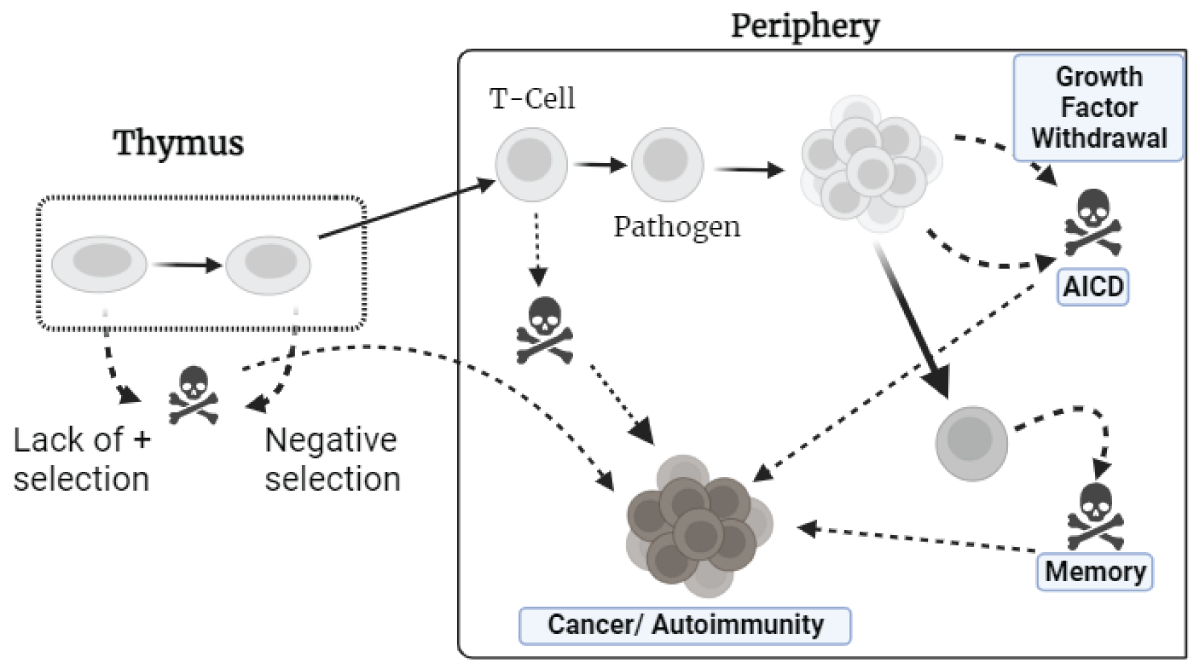
Figure 4: T-cells encounter life-or-death decisions during growth and maturation.
Cell death is crucial in immunogenetics and the resolution of inflammatory responses. Methods and mediators contiguous have yet to be totally understood; however, there is evidence that surrounded non-inflamed cells die via apoptosis, and increase harm produced through inflammatory responses. Figure 5 depicts graphically the several biological processes that mediate and control the inflammatory reaction, as well as the function of apoptosis [25].
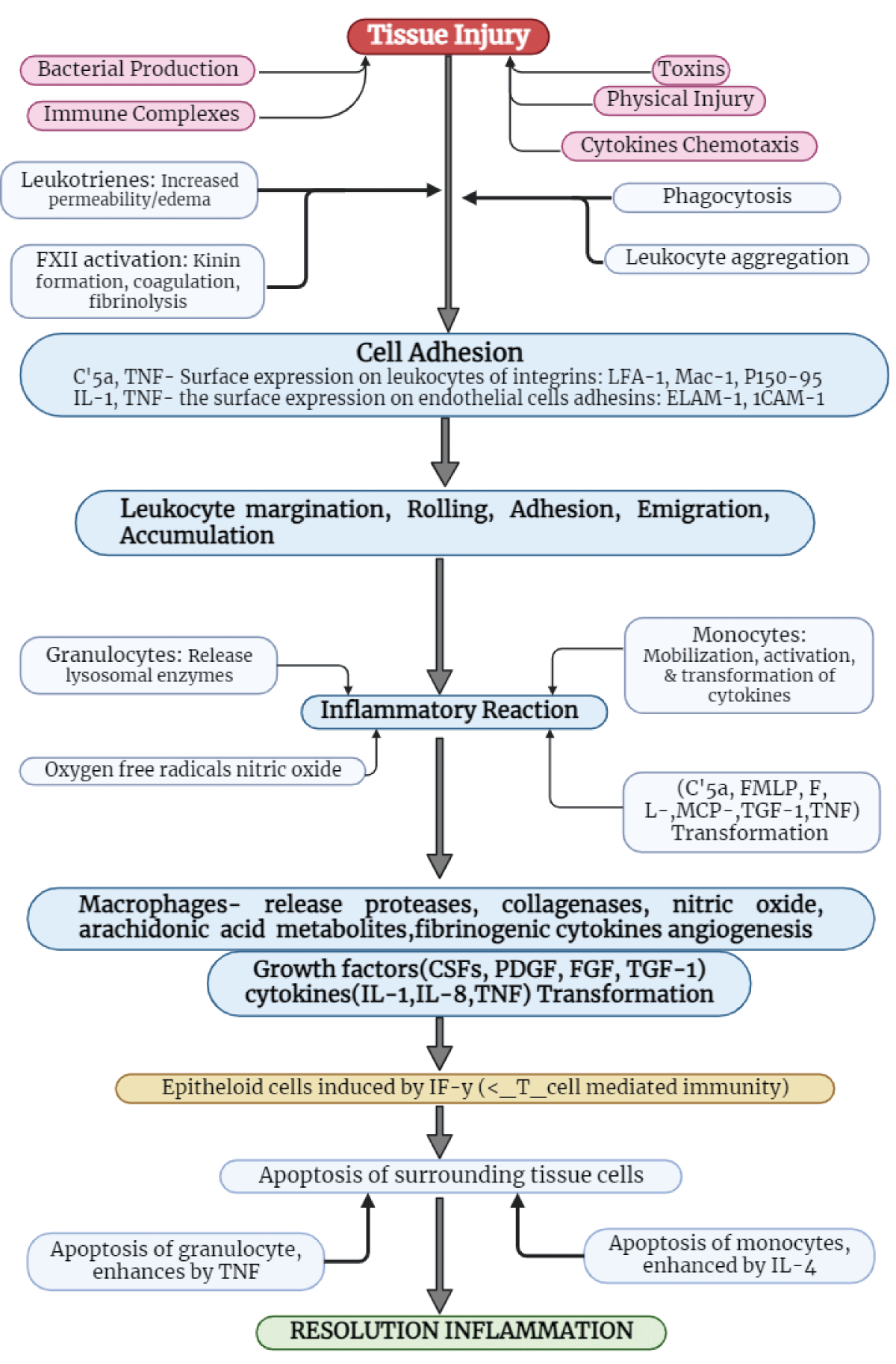
Figure 5: Role of granulocytes, as well as monocytes in apoptosis/inflammation.
Diabetes retinopathy, nephropathy, and diabetic wounds have all been linked to growth factor involvement [26]. Electron microscopy results show the following morphological aspects of cell lysis:
- Cell squeeze
- Chromatin inspissate
- Cytoplasm blister and production of apoptotic carcass
- Phagocytosis of apoptotic entities.
Caspases: Apoptotic machinery’s central effectors. The kin of caspase cysteine proteases plays an important role in cell lysis. So far, 14 caspases have been known, in mammals. The anatomical and operative differences between “initiator, effectors” caspases are reflected in their names. The extended N-terminal prodomain of the active caspase 2, 8, 9, and 10 which in mammals included allows for zymogen clustering and auto-activation. Adaptor proteins, which rely on homophilic interplay with lodge nonenzymatic holding on adaptor and caspase, are required for initiator caspase clustering. FADD, which recruits caspase 8 via a homophilic lysis effectors estate (Figure 2), and Apaf-1, caspase 9 via a homophilic interplay appropriately in the Enlistment domain, are two adaptor molecules that allow initiator caspase auto-activation (CARD). Although different initiator caspases are recruited and activated in different cellular compartments in response to different stimuli (Figure 2), the structural underpinnings of this process bear a striking resemblance to other interacting patterns (death domains, DEDs, CARD). Dimerization is likely employed for auto-activation by effectors caspases downstream (caspases 3, 6, 7) after induced proximity, a strategy used by adaptors to cluster initiator caspases. Caspase 2 is also an “apical” initiator caspase that occurs downstream of intrinsic stress responses and extrinsic death receptor signaling (i.e., before other effector caspases). The splitting of a variety of cellular points by effectors caspases results in the hallmark features of apoptosis. In protest to creator caspases, which are activated prior to cleavage, effector caspases must be ruptured in order to be active. Effectors caspases’ cellular substrates include other caspases, structural, regulatory, and pro-apoptotic proteins [11]. Following the activation of death receptors, apoptotic cells activate a cascade, caspase-8 and perchance caspase-10 the very top of the apoptotic fount. Because caspase-8 iso-forms vary so much more than other caspases, some tissues or cells may be able to defend themselves against CD95- or TNF-induced cell death. The cell death machinery’s activators and effectors are closely linked [6]. When the death domain of either CD95 or TNFR-1 oligomerizes, cytosolic adapter proteins can form a death-propel signaling complex. The apoptotic cascade is therefore activated when cell death receptor CD95 physically connects with the cytosolic protease caspase-8 via the adaptor protein FADD [27]. Millions of effector cells must be recruited quickly and precisely in an antigen-driven way for the immune system to respond successfully. The majority of these effector cells must be destroyed once they have been set alive long enough to perform their work, a procedure known as activation-induced cell death [6]. There are ten major caspases, which may be generally classified as inflammatory caspases, effectors or executioners, and initiators (caspases 2, 8, 9, and 10). Chronic disorders will very certainly necessitate the use of marked inhibitors that target certain initiator caspases, such as caspases-1, 8, or 9 [28]. Caspase 8 colocalizations in lactotrophs enhance in diabetic rat pituitaries, but p53 and nuclear Factor kB are more dramatically activated. These findings suggest that normal cell turnover cascades are partially suppressed in diabetic rats’ pituitaries, perchance via XIAP, and that inhibition may be cell-specific. Even, diabetes-related lactotroph death may be caused by activation of the extrinsic apoptotic route, which includes caspase 8 [29]. IAP antagonists are intriguing cancer therapeutic candidates since they can either directly kill cancer cells or augment current chemotherapy. Controlling the Bcl-2/Bax rheostat, which determines apoptosis susceptibility, is another intriguing technique for treating cancer and other illnesses. Small compounds that imitate BH3 domains are already prototype pharmaceuticals, and their structure-activity relationship profile still has an opportunity for development. Furthermore, it is unclear whether Bcl-2 antagonists and BH3 mimics have a selective and all-encompassing effect on various Bcl-2 family members.
Programmed cell lysis and disease
At present, umpteen disease circumstances involve cell lysis, a lack there for study of cell lysis in pathology makes it difficult to discern and ultimately treat illnesses. The part that follows discusses apoptosis detection techniques and therapeutic alternatives, as well as apoptosis in serious disorders. We will just briefly address these problems because a complete examination of each would be outside the scope of this assessment. As a part of the result, we have concentrated entirely on the function of apoptosis in diabetes and the treatment of diabetic wounds.
Heart
Cardiomyocytes die because of ischemia/reperfusion damage and myocardial infarction. Caspase inhibitor therapy has been demonstrated to aid in the induction of cardiomyocyte apoptosis caused by endotoxin in sepsis. The transcription factors p53, NFkB, AP-1,108, and HGF all appear to regulate cardiomyocyte apoptosis.
Alzheimer’s disease
Apoptosis kills entorhinal, hippocampus, and cortical neurons in Alzheimer’s disease, which is also spotted by amyloid plaques and neurofibrillary tangles. Amyloid can induce neuronal apoptosis, and altered presenilins in Alzheimer’s disease can trigger neuronal death as well. According to a study on the Bcl-2 family in Alzheimer’s disease, Bax levels are greater in neurons from patients than in controls.
Multiple sclerosis
Multiple sclerosis, an autoimmune disorder of the CNS, produces demyelination in white matter regions of the brain and spinal cord. T cells which are self-reactive to auto-antigens, such as myelin protein, myelin-conjugated glycoprotein, myelin’s oligodendrocyte glycoprotein, S-100 protein, andproteo-lipid protein, activate, resulting in demyelination.
Acquired immuno-deficiency syndrome
The virus of human immune deficiency can cause apoptosis in CD8 and CD4 T cells waiting in AIDS. Chronic immune stimulation, gp 120/160 ligation to T-cell gatherer, enhanced manufacture of cytotoxic ligands or viral proteins produced by transmissible cells, direct viral infection, and CD4, T cell death are only a few of the processes that contribute to T-cell depletion. Within the viral proteins that can cause apoptosis are Vpr, Nef, Tat, Vpu, and the Env proteins; they can activate caspase 3 and initiate fas-involved processes. Nef may also reduce Bcl-2 and Bcl-XL levels, disrupting numerous layers of the apoptotic machinery [30]. The Fas/Fas ligand system is involved in the AICD of CD4+ T cells, but it does not appear to be required for CD8+ T cell death. Instead, apoptotic signaling via TNFR is most likely active in CD8+ cells’ AICD. Another research claims that apoptosis contributes to the diabetic nephropathy evolution and the plasma Fas levels may be used to predict outcomes [31].
Role of apoptosis in diabetes mellitus
The global prevalence of diabetes affects over 500 million individuals and in the upcoming 3 decades projected to surpass 1.3 billion. Diabetes mellitus, among the most prevalent non-communicable diseases, affects numerous organ systems and can have catastrophic, life-threatening implications if left untreated. Furthermore, chronic hyperglycemia activates several metabolic processes that result in the toxicity of glucose, which leads to the death of cells [32].
Apoptosis and auto-immunity in diabetes mellitus (Type-1)
Diabetes Type-1 is an illness, albeit accounting for a tiny percentage of the total diabetes population. Pathogenic lymphocyte infiltration of islets starts before clinical indications that indicate enhanced decrement of pancreatic cells of beta-produced autoimmunity by the cell-mediated process. Both T cells as cytotoxic (CD8+) and helper (CD4+) play an active role and are regulated by MHC loci in addition to non-MHC determinants. The associated connection between Type-1 diabetes and an overactive immune system is stronger that T cell-deletion non-obesity diabetic mice do not progress to the illness. When T cells are adopted from a deceased donor and given to a healthy recipient, the disease is also conveyed. Even though the link between cell apoptosis and autoimmunity has not been thoroughly settled, there is growing proof that the T cell-triggered lysis is a prominent effecter approach in diabetes Type-1. β-cells (pancreatic)from recently diagnosed diabetes Type 1 individuals were shown to exhibit enhanced Fas(CD95) on their cellular surfaces than -cells from individuals without underlying health conditions, which do not express detectable Fas (CD95). The apoptotic indication is subsequently transmitted by Fas-ligand conveyed to the invading T-cells. Diabetes develops because of apoptotic cell death; sooner initiation and greater impact in cell lysis result in early illness onset and potentially severe clinical manifestations, whereas later in-life apoptosis (i.e., from the metabolic receiver) results in late diabetes characterized by a slower sledding pattern (Type 2). By combining moderate pathophysiological causes, late-onset type 1 and type 2 diabetes cases may entail moderate-cell lysis [33]. In another study, rats with streptozotocin-induced diabetic neuropathy (DN) displayed abnormalities in their morphological organization, oxidative stress indicators, inflammation cytokines, and apoptosis. Apoptosis may have a role in the development of DN in rats [34]. It has been proposed that apoptosis aids in the course of diabetic nephropathy and plasma sFas power to predict how the disease would progress [31].
Apoptosis and shortage of insulin in Type-2 diabetes mellitus
Diabetes Type-2 is the most frequent kind, defined by a later onset of insulin resistance and/or insufficiency, and amyloidosis. Diabetes Type-1 frequently starts in childhood and has an autoimmune aspect that leads to severe insulin deficiency in the blood. Regardless of the cause or clinical manifestation, hyperglycemia represents the highly prevalent abnormality of the metabolic process in diabetes patients. On the molecular component, pancreatic cell death from apoptosis represents a substantial impact on the onset and/or progression of the illness, as well as the development of insulin insufficiency. In this case, Akt/PKB controls pancreatic cell bulk, purpose of works, and metabolism of carbohydrates through insulin-dependent glucose absorption. The upregulating activity of Ak significantly boosts the resilience process and mutes’ death signals in a multitude of -cell types, whereas down-regulating Akt activity increases the intracellular presented milieu ideal for execution of death [35]. The after-was all reported to be the loss and dysfunction of insulin-releasing cells connected to diabetes mellitus. Diabetes mellitus pathophysiology has been found to include an increased rate of apoptosis; the PI3K/Akt course is critical in determining cell fortune decisions. Insulin insufficiency can be caused by decreased overall cell mass and/or reduced insulin secretion per cell. Despite that glucose intolerance and diabetes are linked to abnormalities in both stages of insulin production. Cross-sectional studies with sufficient control subjects and sample sizes, on the other hand, have given solid evidence for the function of -cell loss in the emergence of diabetes type 2. When compared to non-diabetic control groups, lean and obese type 2 diabetes patients have 10-fold and 3-fold higher rates of -cell apoptosis, respectively (p ≤ 0.05). Whenever compared with non-diabetic age- and weight-matched counterparts, presumed cell mass is lower in both obese and lean people with diabetes type 2. This one result is due to a decrease in the relative volume of cells. In a study, this drop was found independent of how the patients were managed--insulin, sulfonylureas, or diet. The fact that those with IFG have a reduced relative cell volume provides more evidence that this is an early phase and mechanistically crucial in the advancement of diabetes type 2. Finally, the pace of new islet production remained constant, and the decrease in cell mass was due to an increase in the frequency of cell apoptosis. According to the implication, the most rational type 2 diabetes prevention approaches reduce the growing frequency of cell death. Furthermore, while islet neogenesis appears unaffected, inhibiting this 3- to 10-fold higher rate of apoptosis may result in the restoration of -cellular mass in diabetes type 2 patients who have already developed the disease [36]. Indeed, in beta cell physiology, Akt plays a role in regulating the secretion of insulin, volume of cells, neogenesis, and replication, in addition to apoptosis. The proficiency of β-cells to respond to insulin obstruction also a danger factor for diabetes type 2, is required to maintain glucose homeostasis. The substrate of the receptor insulin (IRS-2/PI3K) stream is required to govern -cellular mass and their works. The serine-threonine kinase-Akt, protein kinase B which is the main down pathway target of PI3K process is unfavorably controlled by the phosphatase and tensin homolog deletion at chromosome 10 (PTEN). This Akt signaling process newly has been connected to the pancreatic survival of cells and development through the cell cycle. The IRS2/PI3K/Akt signaling stream has a major impact on β-cell bulk and its function. FoxO1 and GSK3 play important roles in modulating proliferative and survival signals when Akt is active. The Tsc2/mTOR pathway is also important in modulating cell size and proliferation. As a result, we may conclude that no one down the Akt pathway dominates cellular mass; moreover, the individual component is involved in the almost phenotype caused by the activation of Akt. In spite of that, the molecular processes underlying the differences in proliferation and apoptosis caused by various downstream Akt targets must be fully understood. Experiments in mice with the selective removal of specific Akt signaling stream components allow the comprehension of the value of those components in modulating cellular mass [37]. Because environmental factors contribute to the impact of the development of T1D, transfecting cells with the Casp3 gene or using pan-caspase inhibitor therapy may be a way to avoid autoimmune cell destruction for therapeutic purposes. T1D concordance among identical twins is roughly 50%. Prudent glucose control halts the progression of cell apoptosis caused by hyperglycemia.
Pathophysiology associated with healing of diabetic wound and apoptosis
Clinical failures in the healing of wounds pose a challenging issue with serious consequences for morbidity and death in specific patient populations. Wound healing needs sudden spikes in certain cell clusters to mature the wound area, submit a new pattern of matrices, and prepare the wounded area for repair. Before proceeding to the next stage of healing, these particular cell types need to be eradicated from the wound once they have completed their function. The prime kind of cellular diminishment is apoptosis. This condition may remove whole cell clusters without producing tissue impairment or an inflammatory response. The ultimate state observed in excellent wound healing is primarily dependent on fibroblasts. If the fibroblasts are not activated or migrate into the temporary matrix adequately, there will be less autolytic debridement of the denatured proteins and fibrin clot. Fibroblasts serve critical roles in the development and contraction of ECM during wound healing, as well as in the control and regulation of their activity, according to research. Careful management allows for effective wound healing by creating granulation tissue and regulating wound contraction [38]. The particular mechanism by which hyperglycemia affects and decreases wound healing is uncertain. High blood sugar levels have been joined with slow healing of wounds, likewise chronic inflammation, prolonged re-epithelialization, and chronic stress of oxidative condition. Nesfatin-1 (2g/kg/day) therapy enhanced surgical wound healing in rats with normoglycemia and hyperglycemia, while also suppressing apoptosis and stress conditions in the oxidative phase in the dermal and reducing levels of plasma of the inflammation proteins IL-1 and Interleukin-6 (IL-6) [39]. This shows that antioxidant nesfatin-1, anti-inflammatory, and antiapoptotic capabilities may have a major influence on wound healing [39]. Hyperglycemia can also create active oxygen species (ROS) through the AGE, hexosamine, polyol, and protein kinase C course. It is well observed that ROS is needed for the initial stages in the healing of wounds. Disturbances in ROS generation have a deleterious influence on the last phases of wound healing. Increased ROS concentrations can directly affect the metabolism, blood flow, and structural integrity of peripheral neurons. This can cause sensory, motor, and/or autonomic dysfunction in the nerves involved, and each impairment raises the likelihood of developing a Diabetic Foot Ulcer (DFU) in a specific way [40].
The more primitive evolutionary caspase activation mechanism is based on intracellular stress or damage signals conveyed to the Bcl-2 kin members of a vast family that either support or prevent an “intrinsic death program” [27]. Bcl-2 may reduce apoptosis by blocking the translocation of kinase into the cell nucleus [41]. Poor healing of wounds under offloading conditions would be a prominent barrier for human-crewed space expansion missions [42]. Although the approach from which phosphatidylserine (PS)-1 is produced in apoptosis is unknown, PS is actively conveyed from the outside to the innermost leaflet by the aminophospholipid translocase. Because the PS aspect is paired with both loss of aminophospholipid translocase function and increased nonspecific trans-bi-layer translocation of phospholipids, it is suggested that PS may become discernible in the outer leaflet because of the combination of these two events [43]. It is uncertain whether cell cycle stopovers are taken as sensory cues to activate a lethal course, a fact in mitotic catastrophe, or whether apoptosis arises as a direct consequence of alterations in cell cycling component functioning. The connection between the cycling and death of cells should ease the withdrawal of cells incapable of controlling the cell cycle in either situation. As apoptotic pathways may be altered, researchers must exercise alertness when investigating apoptosis in changed cell lines which inherently lack proper cell cycle regulation [41].
Clinical hurdle associated with diabetic wounds and ulcers in the foot
Diabetes has an impact on 422 million persons globally, involving 29.1 million Americans, or 9.3% of the overall total population. Poor or delayed wound healing is a major issue in diabetics, and as a result, persistent non-healing wounds represent significant primary problems associated with diabetes cases. It is expected to affect 15% of cases of diabetes individuals which accounts for an increase to 27% of the $176 billion annually diabetic care cost in the US. Diabetes Foot Ulcers (DFUs) are a particularly critical and difficult kind of diabetes lesion [44]. DFUs are expected to occur at a rate of 6.3% in diabetics globally each year. DFUs contribute to over 60% of lower-limb amputations not due to trauma in the US annually, and individuals undergoing amputation have a mere 40% chance of surviving five years post-procedure. With approximately 1 out of 3 US people developing diabetes by the year 2050 is predicted to reach, the burden and expenses linked with chronic diabetic wounds are predicted to skyrocket, driving the search for innovative therapies [45]. Fibroblasts play an important role in the healing of normal wounds by breaking down the fibrin clot, producing a new extracellular matrix and collagen fabrication to hold the other cells required in the efficient healing of wounds [38]. In hypoxic conditions, heightened necrotic debris accumulation promotes bacterial proliferation, concomitant with compromised immune responses, thus exacerbating microbial challenges. As a result, wounds caused by peripheral vascular disease, as well as conditions about vascular deterioration may be influential, particularly in the case of a diabetic ulcer, and require specific attention. This is seen in a report of older individuals with pressurized ulcers, where the variation in the area of ulcer is highly connected with protein consumption; nevertheless, alternative variables include as vitamins A and C, as well as elemental Zn, are also relevant. However, the delicate balance of these nutrients must be considered. For example, contradicting research on vitamin E, a key lipophilic antioxidant, has demonstrated lower tensile strength and collagen content in experimental wounds [46].
For example, contradicting research on vitamin E, a key lipophilic antioxidant, has demonstrated decreased tensile strength and collagen content in experimental wounds [47].
Another research indicated that decreased cell proliferation, delayed beginning from the myofibroblast phenotype, decreased procollagen-I mRNA manifestation, and abnormal management of cell’s apoptotic lysis may all take part in the poor healing of wound reported in the diabetes paradigm [48].
The act of apoptosis in the healing of wound regulation is explored. Some facts show that apoptosis plays a part in the resolution of various stages of tissue healing. It has been shown that inflammatory cells undergo the onset of apoptosis detection within a 12-hour timeframe trauma in the premature stages of healing tissues. In mice without diabetes and those with diabetes, researchers investigated an exposed covered dermal wound with a dressing of polyurethane. The intriguing discovery is about the constant configuration of inflammatory cell death below the forefront of the migrating epithelium. Granulation of tissues directly adjacent to the wound margin and shielded by an epithelium lacked apoptosis marking and included fewer inflammatory cells and more fibroblasts. Inflammatory cells such as neutrophils, macrophages, and lymphocytes populate the exposed granulation tissue in the middle of the wound. Apoptosis is located at the boundary zone, which is underneath the place at the forefront of the epithelium moving over the tissue’s granulation; however, this study also implies that impairment in inflammatory cell clearance may also contribute to this process [49].
When miR-126 is inhibited, leukocyte adhesion to endothelial cells is said to increase, fostering the healing of wounds. Despite that, nanotechnology is assisting in the advancement of microRNA-based treatment approaches with positive outcomes and few adverse effects [50]. Another relevant study found that DNA fragmentation among cohorts I, II, and III is 40.00 2.97, 45.26 3.21, and 60.8 3.13 (P0.01). Blood sugar levels, particularly postprandial blood sugar (P0.05), are observed to be roughly linearly associated with DNA fragmentation. Proteinuria and serum LDL levels exhibit a significant correlation, while DNA fragmentation is notably higher in the oral hypoglycemic agent (OHA) group compared to the insulin-treated group (p < 0.05). Same, among diabetics with a history of retinopathy, fragmentation of DNA measures 46.50 3.42 (n = 3) in the insulin-treated group and 66.70 6.48 (n = 4) in the OHA group (p < 0.05). This elevated DNA fragmentation notably exacerbates apoptosis in wounds of diabetic characterized by low-regulated levels of blood sugar and microvascular dysfunction. Individuals on OHAs experience this spike more than insulin users, and it inhibits wound healing. Glycemic regulation is essential for avoiding apoptosis and facilitating healing, and insulin outperforms OHA in this regard [51]. As a result, it may be said that strict management of hyperglycemia is necessary for the diabetic lesion to heal.
Wound healing in diabetic versus non-diabetic animals
The three basic stages in wound healing historically as recognized are inflammation, proliferation, with third one remodeling. The inflammatory phase is defined by hemostasis and inflammation. If the streptococcus is present, the incision will not heal, regardless of whether a flap is closed, a skin graft is placed, or primary sutures are utilized. The bacteria impede epithelialization, shortening, and collagen formation while also prolonging the inflammatory phase. Endotoxins cause phagocytosis (apoptosis) and the secretion of collagenase, which assists in collagen breakdown and the obliteration of periphery healthy cells and tissues.
When collagen is exposed during the creation of a wound, the clotting cascade is activated via both intrinsic and extrinsic routes, thereby initiating the inflammation phase. The phase of the inflammatory healing process is delayed by foreign objects and local wound infections (Figure 6). When wound closure is observed, non-diabetic animals heal quickly, with more than 80% recovery within 10 days followed by complete healing within 21 days. The diabetic animal exhibited a noteworthy postponement in wound closure, and the size of the incision did not diminish after 14 days. The wound began to mend after two weeks, and 60% of it had healed by four weeks following the accident. These discoveries align with earlier research [50]. Apoptosis is critical for orchestrating the continually changing cell populations necessary for tissue healing. For the great majority of wounds, proliferation controls result in fast closure. Rarely, the balance between rising and falling cellular populations is disrupted, resulting in pathologic tissue repair. Clinicians may one day design medicines for a wide range of pathologic disorders, including fibrosis; by grasping not just the biological processes that encourage growth but also those that inhibit cell proliferation [52]. Obese and lean people with type 2 diabetes have reduced relative-cell volume and, as a result, lower putative-cell mass when compared to non-diabetic age and weight-matched contemporaries. In the current study, this drop was found independent of how the patients were managed-insulin, sulfonylureas, or diet. Furthermore, those with IFG have lower relative cell volumes, indicating that this is an early process that is critical for diabetes type-2 development [36]. Component of development BCL-2 is an associate in the BCL-2 family. When the PI3K/Akt signaling process is engaged, BAD is phosphorylated, which inhibits apoptosis and promotes cell survival. Akt phosphorylates BAD in vivo and in vitro, and it inhibits BAD-induced primary neuron death in a site-specific manner [35].
Figure 6: Diseases associated with inflammation: When collagen is exposed during the creation of a wound, the clotting cascade is activated via both intrinsic and extrinsic routes, thereby initiating the inflammation phase. The phase of inflammatory healing process is delayed by foreign objects and local wound infections
Limitations of current therapy of diabetic and diabetic wound healing
- Understanding not just the type of cytokines to target, but also when to target them to modify healing, is a vital step in this process [53].
- Anti-angiogenic medications have therefore been proposed as new therapeutics for hypertrophic scars or even keloids, albeit the precise underlying mechanism remains unknown [54].
- Further research is needed to sort out the role of apoptosis in the overall etiology of heart failure, as well as the link between cardiac hypertrophic signaling and apoptosis [20].
- It is not clear how low-molecular-weight compounds were employed to either suppress or improve apoptosis [1].
- With the vast quantity of information we now know regarding cell lysis, our next step is to try translating this understanding techniques of human illness therapy. Another research gap is whether we can cause apoptosis in tumor cells. Do we prevent immune cell apoptosis in AIDS or death by neurological diseases? [30].
- The dynamic nature of the process, detecting apoptosis is difficult. First, apoptosis and the removal of cellular debris can be completed in a matter of hours; evidence of apoptosis may go undetected depending on the sampling time. This may lead to the incorrect notion that apoptosis does not occur and so does not contribute to disease etiology. Furthermore, current methods for detecting apoptosis have technical limitations [15].
Apoptosis variable protein as targets for drug innovation
- Cytokines are essential signaling molecules that are required for recovery or regeneration of cells (Figure 7). More than 30 cytokines are generated through macrophages, fibroblasts, platelets, neutrophils, and epidermal cells during wound healing. Future skin scar research is underway, with a focus on the impact of TGF-b and several variants that can either slow or speed recovery [53].
- Despite diminished angiogenesis, angiotensin is nonetheless required for the proper healing of skin wounds. Despite less angiogenesis than adult skin, lesions in fetal skin and the oral mucosa heal without scarring [54].
- Prostaglandins (PGs) and PGs inhibitors on healing of wound
- Prostaglandins (PGs) are fatty acid molecules that mostly play roles in several physiological and pathologic processes. Three enzymatic transformations are required for prostaglandin production, starting with the conversion of phospholipids derived from the membrane into arachidonic acid through the action of phospholipase. PGE2 is a significant intermediary of inflammation-12 and is implicated in a variety of illnesses, including both types of arthritis as rheumatoid and osteoarthritis. The pathway associated with cyclooxygenase (COX) is required for the transformation of arachidonic acid into PGH2, which is an antecedent of several physiologically active intermediators such as PGE2, thromboxane A2, and prostacyclin. COX-1 (housekeeping gene) has been shown to be presented inherently in different organs, involving the stomach and as induction geneCOX-2, has been found to be triggered through cytokines, tumor promoters, growth factors, and other causing substances. NO is thought to act as a significant disruptor in the wound-healing process. Excess PGE2 derived by COX-2 production plays a critical in-vivo component that accelerates inflammatory response. PGE2 is linked to the proliferation of keratinocytes, the formation of new blood vessels, and inflammatory mediation response. PGE2-associated receptors are categorized as EP1, EP2, EP3, and EP4. PGE2 stimulates intracellular cAMP production by connecting to G-proteins via EP2 and EP4. TGF-b1 was also shown to reduce endogenous COX-2 and PGE2 production within cutaneous fibroblasts, while extraneous PGE2 could counteract TGF-b1-induced collagen-heightened expression mediated by cAMP [55].
Apoptosis has been observed to be induced by nonsteroidal anti-inflammatory medications (NSAIDs) within various cell types [55]. - Eicosanoids
- Prostaglandins, thromboxanes, prostacyclin, lipoxins, and leukotrienes are among the physiologically active, oxygenated arachidonic acid metabolites known as eicosanoids. By using cytochrome P-450 or lipoxygenase, and cyclooxygenase, arachidonic acid can undergo transformation into physiologically active molecules. The arachidonic acid oxygenation from cytochrome P-450 results in the synthesis of epoxy eicosatrienoic acids, and their resultant diols, mono-, di- and tri-hydroxy eicosatetraenoic acids, and derivatives of mono-oxyatedarachidonic acid. The quantity of lipoxin generated has an inverse connection with the formed amount of leukotriene, implying that lipoxins are internal inhibitors that regulate leukotriene activity [12].
- Lymphokines, monokines and interleukins
- Lymphokines refer to cytokines released by activated T cells, while monokines are cytokines secreted by mononuclear phagocytes. Interleukins, a subtype of cytokines, are produced by one type of leukocyte and exert their effects on another leukocyte.
- Colony-stimulating factors
- Colony-stimulating factors are cytokines having wound healing stimulatory action. CSF-1 is a macrophage-released autocrine mediator that "aids in self-preservation. When stimulated, macrophages secrete granulocyte-macrophage CSF that possesses chemotactic and cellular pro-liberation, and in-build activation capabilities [56].
- Interferons
- Interferons (IFN) significantly play a role in the healing of wounds. T-helper lymphocytes are the primary producers, but natural killer and cytotoxic T-cells also contribute. IFN increases the presentation of monocyte antigen and synthesis of cytokine, as well as monocyte effector activities such as adhesion, the process of phagocytosis, secretion patterns, burst in respiratory, and generation of nitric oxide. As a result, macrophages accumulate and intracellular pathogens are destroyed.
- Nitric oxide
- NO is a minuscule radical generated from the L-arginine (amino acid) by 03 different isoforms of Nitric Oxide Synthase (NOS). Calcium concentrations intracellularly are the prime activation mechanisms, resulting in low-level NO generation in a matter of minutes [57].
- Inhibitors of apoptosis
- Hepatitis C Virus infections (HCV) are still occurring. The majority of HCV infections progress to chronicity, culminating in liver damage and failure. HCV accounts for 20% of cases of acute hepatitis, 70% of chronic hepatitis, 40% of end-stage cirrhosis, 60% of liver cell cancer, and 70% of transplantation in liver procedures in developed nations. HCV infects around 200 million individuals globally, with 5 million counts in the United States. The IDN-6556 program aimed at individuals who did not respond to HCV therapy (around a million instances within the United States) [58].
- Other
- Morin's positive effects on cerebral ischemia assault may be due to reduced stress due to oxidative process, suppression of apoptosis, and inflammatory response. The neuroprotective benefits of morin’s supplementation may act as a powerful booster in the treatment of ischemic stroke [58]. Another study reveals that lipid emulsion reduces the bupivacaine-induced rise in the bax/bcl-2 expression ratio; thus, it would be more appropriate to compare the bax/bcl-2 ratio in malathion alone and combination treatment groups with malathion and lipid emulsion [59].
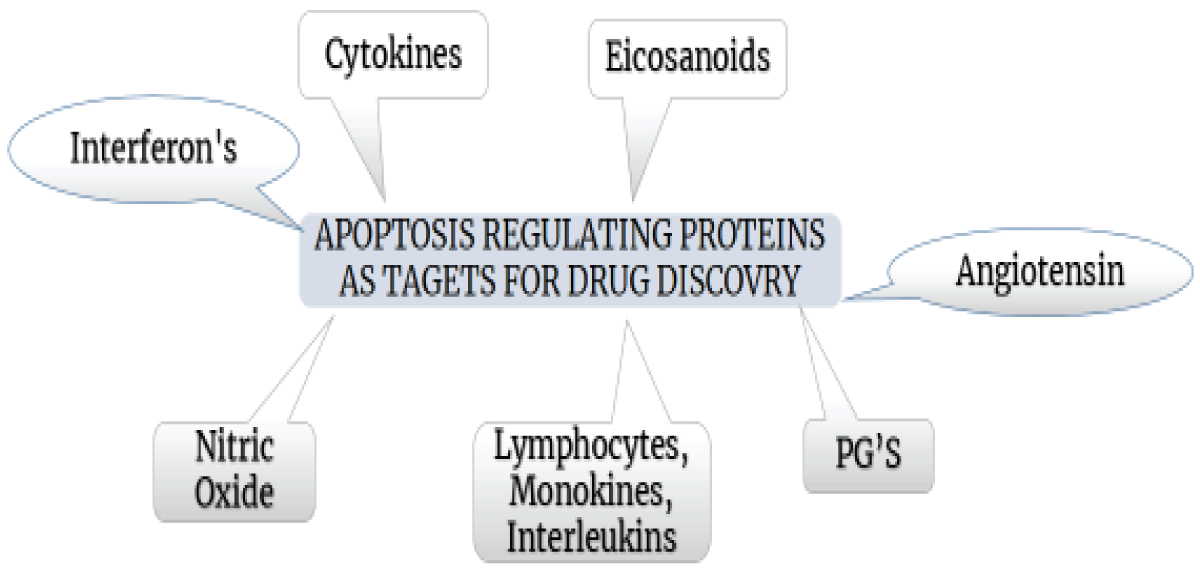
Figure 7: Various proteins that regulate apoptosis as drug discovery targets.
Control of apoptosis can improve the situation: Future treatment strategies
Various medical methodologies and treatment strategies may have an impact on the various mechanisms engaged in the cascade of wound healing. In instances, where the tissue injury is reduced, such as during minimally invasive surgery, the healing time may be quicker, resulting in less soft tissue damage and postoperative morbidity. By regulating inflammation and increasing the proliferative phase, novel approaches of topically growth factor administration and pre-treating with PDGF or IL-1 can enhance the cellular and molecular environment, optimizing it for further processes, hence reducing healing time. Stimulation of the electrical field could enhance the remodeling phase by facilitating better recruitment of fibroblasts and deposition of collagen. Additionally, prosthetic materials might aid in tissue repair, along with potential benefits from gene therapy, which presently in pre-clinical scale-up, could offer a means for targeted healing [60].
Much recent research on the healing of wounds, focused on the modulation of cytokine and chemokine. More focus has been directed to the role of TGF-b isomer and TGF-b itself, as well as the differences in their impact on healing [52].
Stem cell-based treatments have been shown to be effective in both experimental and clinical disorders for skin regeneration and anti-fibrotic characteristics. The human amniotic membrane (HAM) engenders from the innermost strata of the fetus membrane, emerging by the epiblast approximately 8 days post-fertilization, preceding gastrulation. The amniotic membrane contains at least two types of stem cells: Amniotic Epithelial (AECs) and Mesenchymal Cells (AMCs) are two types of stem cells able to regenerate themselves and differentiate into diverse lineages of cells. AECs might be used as a novel anti-fibrotic therapy technique, such as diminishing inflammation at the wound site and orchestrating the transformation of nearby cells to bolster tissue regeneration while thwarting fibrosis [61]. Flow cytometry has emerged as the leading instrumentation/methodology in cell necro-biology. Its applicability spans two areas: one linked to the biology of cell death, and the other as a handy tool for identifying and quantifying apoptotic cells. Laser scanning cytometry (LSC) has similar capabilities to flow cytometry and may be used in place of the latter in most situations. This article provides examples of LSC applications, especially for identifying apoptotic cells. The majority of the methods described were adopted from flow cytometry with slight modifications.
The three-dimensional structures and molecular mechanisms of apoptosis-regulatory proteins are being better understood, leading to strategies for pharmacological intervention in a variety of illnesses discovering the equilibrium within cellular life and death is thrown off, such as in cancer, autoimmune diseases, immunodeficiency, inflammatory, and heart disease (ischemic), diseases related to neurodegenerative and stroke. This manuscript, firstly concentrating on tiny molecule blockers targeting apoptosis-related proteins, described the apoptosis mechanism in its various forms, the relationship between wound and diabetes, the management of programmed cell death within the context of diabetic wound repair, future treatment approaches, and associated research gaps in this field. A variety of additional possible applications for apoptosis-based technologies have not been addressed. Continued research in this captivating field will pave the way for enhanced comprehension regarding the apoptotic system and its regulatory mechanisms.
Declarations
Authors’ contributions: J.P. performed the concept-based study, and supervised scholars; V.D. and M.B. prepared the manuscript and analyzed data; V.D. and J.P. prepared the figures, M.B. carried out the input regarding intelligence and re-revised the manuscript related corrections; all have cross-checked the study and accepted the paramount of this manuscript prior to publication process.
The Authors, J.P., V.D., and M.B. thank the Department of Pharmaceutical Sciences, Mohanlal Sukhadia University, Udaipur, Rajasthan for providing support to this work.
- Kinloch RA, Treherne JM, Furness LM, Hajimohamadreza I. The pharmacology of apoptosis. Trends Pharmacol Sci. 1999 Jan;20(1):35-42. Available from: https://doi.org/10.1016/s0165-6147(98)01277-2
- Sanyaolu A, Marinkovic A, Prakash S, Williams M, Dixon Y, Okorie C, et al. Diabetes mellitus: An overview of the types, prevalence, comorbidity, complication, genetics, economic implication, and treatment. World J Meta-Anal. 2023 Jun 18;11(5):134-143. Available from: http://dx.doi.org/10.13105/wjma.v11.i5.134
- Tomic D, Shaw JE, Magliano DJ. The burden and risks of emerging complications of diabetes mellitus. Nat Rev Endocrinol. 2022;18:525–539. Available from: https://doi.org/10.1038/s41574-022-00690-7
- Harding JL, Pavkov ME, Magliano DJ. The burden and risks of emerging complications of diabetes mellitus. Nat Rev Endocrinol. 2019 Oct;15(10):525-539. Available from: https://doi.org/10.1038/s41574-022-00690-7
- Li J, Jiang C, Xia J. The role of programmed cell death in diabetic foot ulcers: Pathophysiology and therapeutic implications. Int Wound J. 2024 Feb;21(2):143-155. Available from: https://doi.org/10.1111/iwj.14399
- Screaton G, Xu XN. T cell life and death signalling via TNF-receptor family members. Curr Opin Immunol. 2000 Jun;12(3):316-322. Available from: https://doi.org/10.1016/s0952-7915(00)00093-5
- Fischer U, Schulze-Osthoff K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005;12:942-961. Available from: https://doi.org/10.1038/sj.cdd.4401556
- Fischer U, Schulze-Osthoff K. New approaches and therapeutics targeting apoptosis in disease. Pharmacol Rev. 2005 Jun;57(2):187-215. Available from: https://doi.org/10.1124/pr.57.2.6
- Talanian RV, Brady KD, Cryns VL. Caspases as targets for anti-inflammatory and anti-apoptotic drug discovery. J Med Chem. 2000 Aug;43(17):3351-3371. Available from: https://doi.org/10.1021/jm000060f
- Schumacher M, Cerella C, Reuter S, Dicato M, Diederich M. Anti-inflammatory, pro-apoptotic, and anti-proliferative effects of a methanolic neem (Azadirachta indica) leaf extract are mediated via modulation of the nuclear factor-κB pathway. Genes Nutr. 2011 Apr;6(2):149-160. Available from: https://doi.org/10.1007/s12263-010-0194-6
- Martin DA, Elkon KB. Mechanisms of apoptosis. Rheum Dis Clin North Am. 2004 May;30(2):441-454. Available from: https://www.academia.edu/12492421/Mechanisms_of_apoptosis
- Kinchen JM, Ravichandran KS. Journey to the grave: Signaling events regulating removal of apoptotic cells. J Cell Sci. 2007 Jul;120(13):2143-2149. Available from: https://doi.org/10.1242/jcs.03463
- Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol. 1999 Apr;11(2):255-260. Available from: https://doi.org/10.1016/s0955-0674(99)80034-9
- Koff JL, Ramachandiran S, Bernal-Mizrachi L. A time to kill: Targeting apoptosis in cancer. Int J Mol Sci. 2015 Feb;16(2):2942-2955. Available from: https://doi.org/10.3390/ijms16022942
- Best PJM. Brief Review. Arterioscler Thromb Vasc Biol. 1999:14-23.
- Liadis N, Murakami K, Eweida M, Elford AR, Sheu L, Gaisano HY, et al. Caspase-3-dependent β-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol. 2005;25(9):3620-3629. Available from: https://doi.org/10.1128/mcb.25.9.3620-3629.2005
- Amaravadi R. Autophagy can contribute to cell death when combining targeted therapy. Cancer Biol Ther. 2009;8:130-133. Available from: https://doi.org/10.4161/cbt.8.21.10416
- Koh TJ, DiPietro LA. Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med. 2011;13:e23. Available from: https://doi.org/10.1017/s1462399411001943
- Martin DA, et al. Apoptosis: Biphasic dose responses. Crit Rev Toxicol. 2001;17.
- Kang PM, Izumo S. Apoptosis in heart: Basic mechanisms and implications in cardiovascular diseases. Trends Mol Med. 2003;9(4):177-182. Available from: https://doi.org/10.1016/s1471-4914(03)00025-x
- Bosman FT, Visser BC, van Oeveren J. Apoptosis: Pathophysiology of programmed cell death. Pathol Res Pract. 1996;192:676-683. Available from: https://doi.org/10.1016/s0344-0338(96)80089-6
- Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17(8):759-771. Available from: https://doi.org/10.1038/cr.2007.52
- Huang NF, Zac-Varghese S, Luke S. Apoptosis in wound healing. Available from: https://www.hmpgloballearningnetwork.com/site/wounds/article/1746
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6(4):232-241. Available from: https://doi.org/10.1038/nrrheum.2010.4
- Haanen C, Vermes I. Apoptosis and inflammation. Mediators Inflamm. 1995;4(1):5-15. Available from: https://doi.org/10.1155/s0962935195000020
- Rai NK, Suryabhan, Ansari M, Kumar M, Shukla VK, Tripathi K. Effect of glycaemic control on apoptosis in diabetic wounds. J Wound Care. 2005;14(7):277-281. Available from: https://doi.org/10.12968/jowc.2005.14.6.26792
- Martin SJ. Caspases: Executioners of Apoptosis. Pathobiology of Hum Dis. 2014;16:145-152. Available from: https://cir.nii.ac.jp/crid/1360855569585741184
- Reed JC. Apoptosis-regulating proteins as targets for drug discovery. Trends Mol Med. 2001;7(8):314-319. Available from: https://doi.org/10.1016/s1471-4914(01)02026-3
- Arroba AI, Frago LM, Argente J, Chowen JA. Activation of caspase 8 in the pituitaries of streptozotocin-induced diabetic rats: Implication in increased apoptosis of lactotrophs. Endocrinology. 2005;146:4417-4424. Available from: https://doi.org/10.1210/en.2005-0517
- Baba K, Minatoguchi S, Sano H, Kagawa T, Murata I, Takemura G, et al. Involvement of apoptosis in patients with diabetic nephropathy: A study on plasma soluble Fas levels and pathological findings. Nephrology. 2004;9:94-99. Available from: https://doi.org/10.1111/j.1440-1797.2004.00238.x
- Tomita T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn J Basic Med Sci. 2016;16(3):162-179. Available from: https://doi.org/10.17305/bjbms.2016.919
- Lee SC, Pervaiz S. Apoptosis in the pathophysiology of diabetes mellitus. Int J Biochem Cell Biol. 2007;39(3):497-504. Available from: https://doi.org/10.1016/j.biocel.2006.09.007
- Sha J, Sui B, Su X, Meng Q, Chenggang Z. Alteration of oxidative stress and inflammatory cytokines induces apoptosis in diabetic nephropathy. Mol Med Rep. 2017;16(6):7715-7723. Available from: https://doi.org/10.3892/mmr.2017.7522
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231-241. Available from: https://doi.org/10.1016/s0092-8674(00)80405-5
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Humans with Type 2 Diabetes. Diabetes. 2003;52(1):102-110. Available from: https://doi.org/10.2337/diabetes.52.1.102
- Elghazi L, Bernal-Mizrachi E. Akt and PTEN: β-cell mass and pancreas plasticity. Trends Endocrinol Metab. 2009;20(5):243-251. Available from: https://doi.org/10.1016/j.tem.2009.03.002
- Xu Y, Chen F. Antioxidant, anti-inflammatory, and anti-apoptotic activities of Nesfatin-1: A review. J Inflamm Res. 2020;13:607-617. Available from: https://doi.org/10.2147/jir.s273446
- Burgess JL, Wyant WA, Abujamra BA, Kirsner RS, Jozic I. Diabetic wound-healing science. Medicina. 2021;57(10):1072. Available from: https://doi.org/10.3390/medicina57101072
- Riwaldt S, Corydon TJ, Pantalone D, Sahana J, Wise P, Wehland M, et al. Role of apoptosis in wound healing and apoptosis alterations in microgravity. Front Bioeng Biotechnol. 2021;9:1-22. Available from: https://doi.org/10.3389/fbioe.2021.679650
- Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the amino phospholipid translocase. J Biol Chem. 1997;272(42):26159-26165. Available from: https://doi.org/10.1074/jbc.272.42.26159
- King KL, Cidlowski JA. Cell cycle and apoptosis: common pathways to life and death. J Cell Biochem. 1995;58(2):175-180. Available from: https://doi.org/10.1002/jcb.240580206
- Kimball AS, Davis FM, Joshi AD, Schaller MA, Bermick J, Xing X, et al. The histone methyltransferase Setdb2 modulates macrophage phenotype and uric acid production in diabetic wound repair. Immunity. 2019;51(2):258-271. Available from: https://doi.org/10.1016/j.immuni.2019.06.015
- Cho H, Blatchley MR, Duh EJ, Gerecht S. Acellular and cellular approaches to improve diabetic wound healing. Adv Drug Deliv Rev. 2019;146:267-288. Available from: https://doi.org/10.1016/j.addr.2018.07.019
- Bainbridge P. Wound healing and the role of fibroblasts. J Wound Care. 2013;22(8):407-408. Available from: https://doi.org/10.12968/jowc.2013.22.8.407
- Han G, Ceilley R. Chronic wound healing: A review of current management and treatments. Adv Ther. 2017;34(6):599-610. Available from: https://doi.org/10.1007/s12325-017-0478-y
- Mutschler W. Physiologie und pathophysiologie der heilung von defektwunden. Unfallchirurg. 2012;115:767-773. Available from: https://doi.org/10.1007/s00113-012-2208-x
- Darby IA, Bisucci T, Hewitson TD, MacLellan DG. Apoptosis is increased in a model of diabetes-impaired wound healing in genetically diabetic mice. Int J Biochem Cell Biol. 1997;29:191-200. Available from: https://doi.org/10.1016/s1357-2725(96)00131-8
- Brown DL, Kao WWY, Greenhalgh DG. Apoptosis down-regulates inflammation under the advancing epithelial wound edge: Delayed patterns in diabetes and improvement with topical growth factors. Surgery. 1997;121:372-380. Available from: https://doi.org/10.1016/s0039-6060(97)90306-8
- Davis FM, Kimball A, Boniakowski A, Gallagher K. Dysfunctional wound healing in diabetic foot ulcers: New crossroads. Curr Diabetes Rep. 2018;18:2. Available from: https://doi.org/10.1007/s11892-018-0970-z
- Rai NK, Ansari M, Kumar M, Shukla VK, Tripathi K. Apoptosis: A basic physiologic process in wound healing. Int J Lower Extrem Wounds. 2005;4(3):138-144. Available from: https://doi.org/10.1177/1534734605280018
- Greenhalgh DG. The role of apoptosis in wound healing. Int J Biochem Cell Biol. 1998;30(11):1019-1030. Available from: https://doi.org/10.1016/s1357-2725(98)00058-2
- Gantwerker EA, Hom DB. Skin: Histology and physiology of wound healing. Facial Plast Surg Clin North Am. 2011;19(3):441-453. Available from: https://doi.org/10.1016/j.fsc.2011.06.009
- Sorg H, Tilkorn DJ, Hager S, Hauser J, Mirastschijski U. Skin wound healing: An update on the current knowledge and concepts. Eur Surg Res. 2017;58(1-2):81-94. Available from: https://doi.org/10.1159/000454919
- Geske FJ, Gerschenson LE. The biology of apoptosis. Hum Pathol. 2001;32(10):1029-1038. Available from: https://doi.org/10.1053/hupa.2001.28250
- Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S. Nonsteroidal anti-inflammatory drugs induce apoptosis in association with activation of peroxisome proliferator-activated receptor-γ in rheumatoid synovial cells. J Pharmacol Exp Ther. 2002;302(1):18-25. Available from: https://doi.org/10.1124/jpet.302.1.18
- Gharagozloo M, Kalantari H, Rezaei A, Maracy MR, Salehi M, Bahador A, et al. Immune-mediated cochleovestibular disease. Bratisl Med J. 2015;116(5):296-301.
- Broughton G, Janis JE, Attinger CE. Wound healing: An overview. Plast Reconstr Surg. 2006;117:1-32. Available from: https://doi.org/10.1097/01.prs.0000222562.60260.f9
- Melnikova I, Golden J. Apoptosis-targeting therapies. Nat Rev Drug Discov. 2004;3(11):905-906. Available from: https://doi.org/10.1038/nrd1554
- Chen Y, Li Y, Xu H, Li G, Ma Y, Pang YJ. Morin mitigates oxidative stress, apoptosis and inflammation in cerebral ischemic rats. Afr J Tradit Complement Altern Med. 2017;14:348-355. Available from: https://doi.org/10.21010/ajtcam.v14i2.36
- Velnar T, Bailey T, Smrkolj V. The wound healing process: An overview of the cellular and molecular mechanisms. J Int Med Res. 2009;37(5):1528-1542. Available from: https://doi.org/10.1177/147323000903700531
- Wang PH, Huang BS, Horng HC, Yeh CC, Chen YJ. Wound healing. J Chin Med Assoc. 2018;81(2):94-101. Available from: https://doi.org/10.1016/j.jcma.2017.11.002
- Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495-516. Available from: https://doi.org/10.1080/01926230701320337