Research Article
Arid3a regulates mesoderm differentiation in mouse embryonic stem cells

Melissa Popowski1,2, Bum-kyu Lee1,2, Cathy Rhee3, Vishwanath R Iyer1,2 and Haley O Tucker1,2*
1Department of Molecular Biosciences, USA
2Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, Texas 78712, USA
3Center for Regenerative Medicine, Massachusetts General Hospital, Boston, MA 02114, USA
*Address for Correspondence: Haley O Tucker, Department of Molecular Biosciences, Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, Texas 78712, USA. Email: HaleyTucker@austin.utexas.edu
Dates: Submitted: 17 August 2017; Approved: 06 September 2017; Published: 07 September 2017
How to cite this article: Popowski M, Lee BK, Rhee C, Iyer VR, Tucker HO. Arid3a regulates mesoderm differentiation in mouse embryonic stem cells. J Stem Cell Ther Transplant. 2017; 1: 052-062. DOI: 10.29328/journal.jsctt.1001005
Copyright License: © 2017 Popowski M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Research into regulation of the differentiation of stem cells is critical to understanding early developmental decisions and later development growth. The transcription factor ARID3A previously was shown to be critical for trophectoderm and hematopoetic development. Expression of ARID3A increases during embryonic differentiation, but the underlying reason remained unclear. Here we show that Arid3a null embryonic stem (ES) cells maintain an undifferentiated gene expression pattern and form teratomas in immune-compromised mice. However, Arid3a null ES cells differentiated in vitro into embryoid bodies (EBs) significantly faster than control ES cells, and the majority forming large cystic embryoid EBs. Analysis of gene expression during this transition indicated that Arid3a nulls differentiated spontaneously into mesoderm and neuroectoderm lineages. While young ARID3A-deficient mice showed no gross tissue morphology, proliferative and structural abnormalities were observed in the kidneys of older null mice. Together these data suggest that ARID3A is not only required hematopoiesis, but is critical for early mesoderm differentiation.
Introduction
Mouse embryonic stem (mES) cells are invaluable research tools that have allowed researchers to unravel the early stages of cell differentiation and lineage commitment [1]. In normal development, pluripotent ES cells quickly expand and become committed to specific cell lineages. Cell-cell interactions and timing of differentiation are critical to the correct formation of the embryo [2]. Understanding the initial cell fate decisions offers insight into early development.
Pluripotent and multipotent cells are the critical reservoirs for functional, fully differentiated cells of all organisms. Arid3a is a transcription factor that is broadly expressed during early development. Its expression becomes restricted first to the fetal liver, and then primarily to B lymphocytes within the adult [3,4]. In the mouse, Arid3a is required for hematopoietic stem cell (HSC) development, which occurs in the fetal liver at E12.5 [4,5], and is a well-established regulator of B-cell development in the adult mouse. We have previously shown that the loss of Arid3a engenders a defect in HSC development during embryogenesis [4], causing nearly all Arid3a null (Arid3a-/-) animals to die by E12.5. Rare survivor mice are typically smaller than either their wildtype (Arid3a+/+) or heterozygous (Arid3a ±) littermates, but grow to full size as they reach maturity. We have further shown that Arid3a protein levels during differentiation of ES cells [6-8] and controls trophectoderm, and subsequent placental development directly via repression of key pluripotency factors [7,9]. However, the function of Arid3a in further ES cell differentiation was unknown. These observations lead us to investigate how Arid3a influences early embryonic developmental choices and subsequent adult tissue development.
Results
To understand the role Arid3a plays in normal mES cells, we utilized Arid3a null embryonic stem cells (Arid3a-/- mES) [4]. These cells maintain mES morphology (Figure 1a) stably through many passages. They grow at equivalent rates compared to Arid3a+/+ and Arid3a± mES cells and showed no abnormal phenotype and maintained expression of pluripotency factors (data not shown and Figure 2b and 3a). The presence of typical mES cell morphology is indicative of the maintenance of pluripotency, however it is inconclusive. In order to test for pluripotency, teratoma assays were performed [9]-a standard criterion for pluripotency. Arid3a-/- mES cells formed teratomas with a similar latency and cellular composition as Arid3a+/+ mES cells, indicating that they maintained pluripotency in culture (Figure 1b).
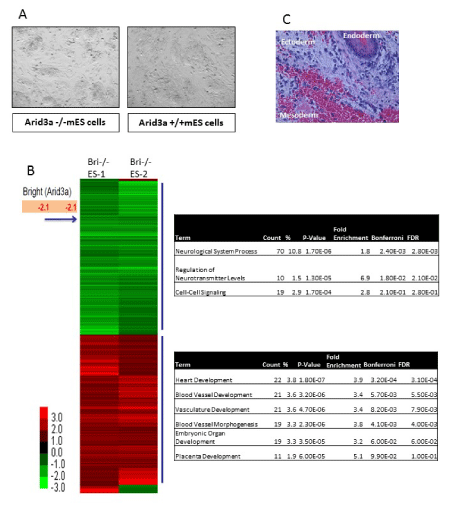
Figure 1: Arid3a knockout embryonic stem cells are pluripotent. (A) Arid3a-/- ES morphologically resemble mES. Arid3a-/- ES cells grown on a feeder cell layer, in the presence of LIF, maintain their ES morphology. (B) Arid3a knockout embryonic stem cells form teratomas. ~350,000 Arid3a-/-ES cells were injected into the flanks of immune compromised mice. Tumors were harvested when ~1cm across. Sections were stain by H&E, and cells representing all three germ layers were found to be present. Abbreviations: Arid3a-/- ES: Arid3a knockout embryonic stem cells; mES: mouse embryonic stem cells. (C) Global gene expression analysis was performed on biological replicates (replicate 1 and replicate 2) of two independently derived Arid3a knockout embryonic stem cell lines (Arid3a-/- ES-1 &-2). Arid3a-/- ES were compared to Arid3a+/+ ES cells. Heatmaps represent the differentially expressed genes between Arid3a-/- ES lines and Arid3a+/+ ES greater than 1-fold difference. Gene ontogeny analyses of differentially expressed genes are displayed to the right of the heatmap. Abbreviations: Arid3a-/- ES: Arid3a knockout embryonic stem cells; Arid3a+/+ ES: wildtype mouse embryonic stem cells.
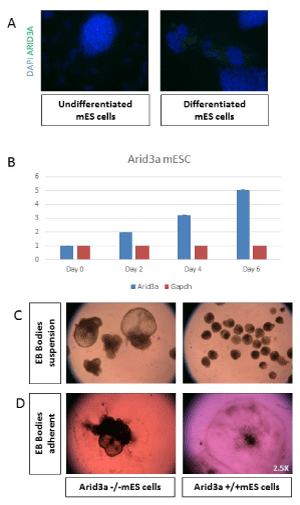
Figure 2: Arid3a increases expression and influences differentiation. (A) mES cells show an increase in Arid3a protein levels upon differentiation. mES cells were grown in chamber slides in the presence (undifferentiated) or absence (differentiated) of LIF for 3 days. Cells were stained for Arid3a expression (green), and nuclei were counterstained with DAPI (blue). (B) Arid3a +/+ mESC show an increase in Arid3a expression during differentiation induced through removal of stem cell factors compared to GAPDH expression. (C) Arid3a -/- mESC differentiate into embryoid bodies more rapidly compared to wildtype. Two different Arid3a-/- ES cell lines (Arid3a-/- ES-1 and Arid3a-/- ES-2) and Arid3a+/+ ES cells were grown in suspension culture without LIF to promote differentiation. Arid3a-/- ES formed CEBs more quickly, more often, and much larger then Arid3a+/+ ES. Images are at day 10 differentiation. 2.5x magnification. (C) Arid3a-/- mES do not differentiate correctly in adherent cultures. Two different Arid3a-/- cell lines and Arid3a+/+ mES cells were grown in suspension culture without LIF to promote formation of EBs for eight days. EBs were allowed to adhere to gelatin coated cell culture plates. Arid3a-/- mES cells did not appropriately adhere to the cell culture plate and appeared to incompletely differentiate. Images are at day 13 differentiation (2.5x magnification).
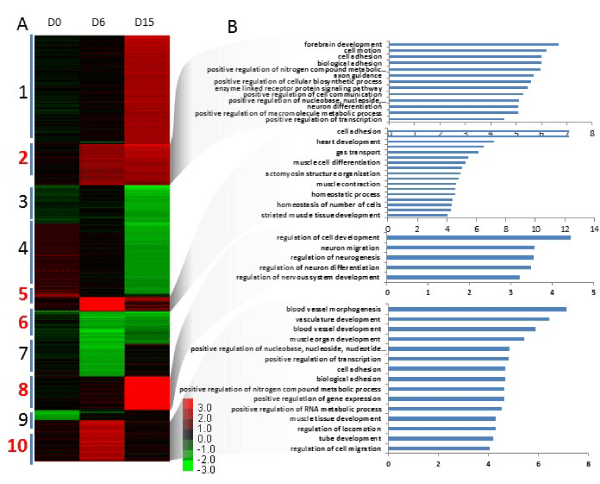
Figure 3a,b: Gene expression of differentiated Arid3a knockout embryonic stem cells is dissimilar to wildtype embryonic stem cells. (A) Global gene expression analysis was performed on biological replicates of two independently derived Arid3a knockout embryonic stem cell lines (Arid3a-/- ES-1 & 2) and Arid3a+/+ ES. Cells were cultured in hanging drops to allow differentiation into embryoid bodies and harvested at days 0, 6, and 15 (D0, D6, and D15, respectively). All Arid3a-/- ES replicates were compared together to Arid3a+/+ ES cells. The heatmap represents the differentially expressed genes (greater the 1.5-fold difference and a P value less than 0.05) clustered based on expressed patterns over time. Data are a compilation between replicates of both Arid3a-/- ES lines and Arid3a+/+ ES at each timepoint. Abbreviations: Arid3a-/- ES: Arid3a knockout embryonic stem cells; Arid3a+/+ ES: wildtype mouse embryonic stem cells. (B) Gene ontogeny analyses of differentially expressed genes in differentiated Arid3a knockout embryonic stem cells. Clusters were analyzed using Gene ontogeny and show a wide range of misregulated gene pathways. Generally, differentiation associated pathways are more commonly upregulated while stem cell related programs are more downregulated.
To compare the genetic expression profile of Arid3a-/- and Arid3a+/+ mES, microarray analysis was performed on undifferentiated Arid3a-/- cells (Figure 1c). All arrays were performed using two independently derived Arid3a-/- mES cell lines (Arid3a-/- mES-1 and Arid3a-/- mES-2) [9]. mES cells were grown on feeder cultures, and microarrays were performed using sorted (via flow cytometry) and unsorted (feeder cells were removed via re-attachment to substrate) biological replicates (Figure 1c, data not shown). We observed few differences between the two arrays. Gene ontogeny (GO) analyses comparing Arid3a-/- to WT mESC revealed several common downregulated pathways, including neurological system processes, regulation of neurotransmitter levels, and cell-cell signaling. Unexpectedly, the upregulated pathways included heart and vasculature- associated development, such as heart, blood vessel, vasculature development and blood vessel morphogenesis (Figure 1c).
Previous differentiation analyses of mES showed that Arid3a was highly upregulated upon differentiation of mES cells [9,10], suggesting a function for Arid3a in both pluripotency and differentiation. Consistent with those observations [9,10], Arid3a in undifferentiated mES was increased during serum withdrawal-mediated differentiation (Figure 2a,b).
Additional in vitro differentiation experiments were performed to explore this issue further. When retained in suspension cultures, mES differentiate to embryoid bodies (EBs).-a round mass of differentiated cells composed of three germ lineages [11,12]. Arid3aefly, cells were removed from feeders and plated in hanging drop cultures for 2 days. On the third day, EBs were collected and grown under suspension conditions for up to 20 days (Figure 2c). The Arid3a-/- CEBs were almost always much larger than any Arid3a+/+ EBs. We observed that Arid3a-/- EBs grew much faster than Arid3a+/+ controls, and the majority formed very large structures termed cystic embryoid bodies (CEBs) [13]. CEBs are models for early extraembryonic tissues development, as they contain yolk-sac-like structures [14,15] and early vasculature [13,16].
To determine if formation of CEBs were progenitors of cardiac differentiation, Arid3a-/- and Arid3a+/+ EBs were plated on gelatin coated plates and cultured according to previous protocols (Figure 2d) [12,17]. Arid3a+/+ EBs plated down and formed regular, round colonies with differentiated cells radiating out from the center of less differentiated cells and were able to form beating cardiomyocytes (data not shown). Although Arid3a-/- EBs formed beating cardiomyocytes, they did not spread out on the plate nor form regular differentiating colonies (Figure 2d). These morphological differences further indicated that Arid3a-/- mES cells were incapable of differentiating efficiently to cardiomyocyte progenitor lineages.
To determine the cellular processes that might underlie these Arid3a-/- mES differentiation defects, we performed gene expression analysis on undifferentiated Arid3a-/- and Arid3a+/+ mES controls as well as on EBs harvested at day 6 and 15. Employing two biological replicates for each time-point, a heat map was generated by averaging all Arid3a-/- replicates for each time-point and normalizing each to the Arid3a+/+ controls. Genes that were >2-fold up- or down-regulated for each time-point were plotted and clustered according to expression patterns over the full time course. There were surprisingly few differences between Arid3a+/+ and -/- mES cells observed at day 0 (Figure 3a). But differences increased at day 6, with the largest gene expression variation observed at day 15 (Figure 3a). GO analysis of select clusters (numbered in red) showed a range of deregulated pathways (Figure 3b). For example, Cluster 6 included downregulation of genes involved in pluripotency for stem cell maintenance, stem cell development, and gastrulation. Also downregulated in Cluster 6 were related differentiation pathways, including stem cell differentiation, formation of the primary germ layer, pattern specification process, and anterior/posterior pattern formation. We also noted that the same pathways deregulated in undifferentiated mES cell microarrays (Figure 1b) were also upregulated in Clusters 2, 5, 8, and 10 (Figure 3b-f). These could roughly be segregated into two groups: Vasculature (heart, blood vessel, vasculature development, and blood vessel morphogenesis) and Adhesion/motility (cell motion, cell adhesion, biological adhesion, cell-cell adhesion, and regulation of cell migration). The upregulation of vasculature-related pathways correlated with the increased formation of CEBs in Arid3a-/- mES cells. We noted previously that Arid3a-/- EBs did not properly adhere to the cell culture plate nor spread-out when plated in hanging drops The increase in adhesion pathways seen from the GO analyses may explain this observation.
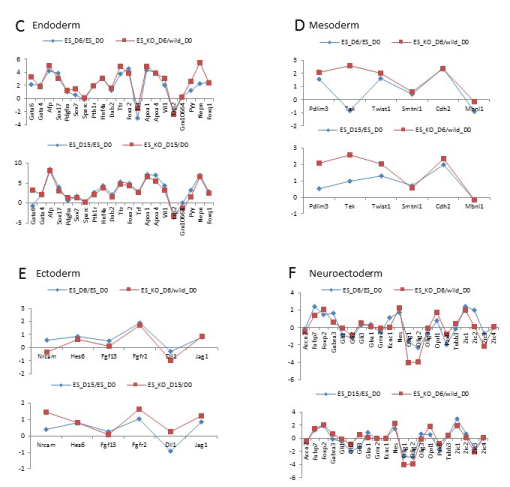
Figure 3c-f: Gene expression of differentiated Arid3a knockout embryonic stem cells is dissimilar to wildtype embryonic stem cells. Arid3a-/- ES differentiate into endoderm and ectoderm lineages in a similar manner to Arid3a+/+ ES. Arid3a-/- ES cells express mesoderm and neuroectoderm markers sooner than the Arid3a+/+ ES cells. Fold expression of all genes relative to Arid3a+/+ ES undifferentiated control (D0).
We next analyzed gene expression data comparing deregulation of lineage-specific genes. Arid3a-/- mES cells differentiated more rapidly down certain lineage pathways (Figure 3c-f). This increased differentiation rate may account for the greater change in gene expression observed at D15 compared to D0. Yet Arid3a-/- mES cells appeared to differentiate at the same pace as controls toward the endoderm lineage (Figure 3c) and followed a similar (but not identical) timing pattern in differentiating to the ectoderm lineage (Figure 3e). However, Arid3a-/- mES differentiated into neuroectoderm and mesoderm lineages more efficiently than wildtype controls (Figure 3d,f). Correlation analysis indicated that neuroectoderm and mesoderm patterns of Arid3a-/- EBs at day 6 EBs more closely resembled the patterns of Arid3a+/+ controls at E15 (Figure 3d,f).
While most Arid3a-/- mice die by E12.5, there are rare survivors [4]. To study the effect of Arid3a loss on organ differentiation, histological analysis was performed on multiple tissue types of young (6 week-old) and aged (>1 year old) Arid3a-/-, Arid3a±, and Arid3a+/+ littermates. Paraffin embedded sections from various organs were stained with H&E and gross anatomical analysis was performed. As shown in Figure 4a, there were no identifiable differences between KOs and controls in formation, size, or cellular composition of the Arid3a-/- organs at 6-weeks. However, in aged mice, while most tissue types were normal, a distinct loss of structure was observed in their kidneys (Figure 4a).
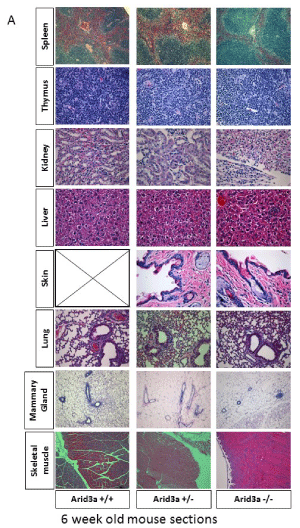
Figure 4a: Arid3 -/- mice show no structural abnormalities in most major organs until aging. (A) H&E staining of Arid3a wildtype (Arid3a+/+), heterozygous (±) and knockout (Arid3a-/-) littermates was performed on the spleen, thymus, kidney, liver, skin, lung, mammary gland, and skeletal muscle. No structural abnormalities were observed. Black box indicates no data available.
As a first step to determine whether early embryonic defects in Arid3a expression influence kidney development, we analyzed gross cellular proliferation in the kidneys and spleens of Arid3a-/- and control littermates (Figure 4b). Histological sections from Arid3a+/+ and Arid3a-/- spleens and kidneys were stained with the proliferative marker Ki67. The analysis of Figure 4c indicated that there is a significant increase in proliferation within the kidney, but not the spleen, of Arid3a-/- young mice, but not in the spleen (Figure 4d). These data indicate that Arid3a influences early differentiation of neuroectoderm and mesoderm germ layers and plays a heretofore unrecognized role in the adult kidney.
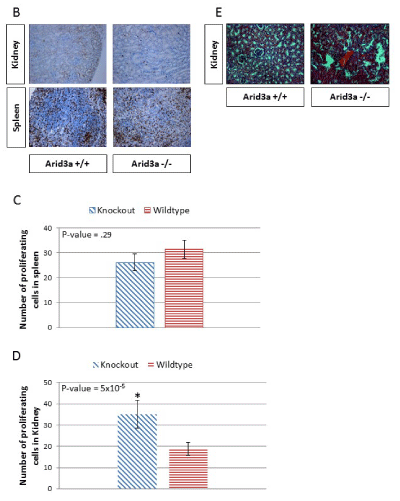
Figure 4b-d: (B) Spleen and kidney of 6 week-old Arid3a wildtype (Arid3a+/+) and knockout (Arid3a-/-) littermates were stained for the proliferative marker Ki67 (brown) and the nuclei counterstained with eosin (blue). (C) The number of proliferating cells in Arid3a wildtype (Arid3a +/+) and knockout (Arid3a-/-) spleens were averaged by randomly counting ten equally sized squares from each stain. The average number of positive cells for each section was averaged from each square counted; variance was determined by F-test and found to be equal. Student’s T-test was performed to determine the P-value. All calculations were performed in Microsoft Excel. (D) The number of proliferating cells in Arid3a wildtype (Arid3a+/+) and knockout (Arid3a -/-) kidney were counted for the entire section. The numbers of positive cells were averaged; the variances were determined by F-test and found to be equal. Student’s T-test was performed to determine P-value. (E) H&E staining of Arid3a wildtype (Arid3a+/+), heterozygous (±) and knockout (Arid3a-/-) age matched mice (> 6 months) was performed on the kidney. Structural abnormalities were observed only the older Arid3a -/- kidneys.
Discussion
Arid3a has multiple cellular functions in addition to as a transcription factor. For instance, Arid3a shuttles between the nucleus and the cytoplasm as well as localizes to lipid rafts to affect B cell receptor signaling [18,19]. Arid3a was shown to interact with cell signaling factors, including p53, to regulate senescence [20-22]. Arid3a transactivation activity is maximal when localized to the nuclear matrix [23] to enhance chromatin accessibility [24]. Previous evidence has shown that Arid3a is essential for embryonic hematopoiesis in the fetal liver as well as normal kidney development in the adult [4,9]. Arid3a is required for maintaining the differentiation state of various somatic cell types, as its sole deficiency leads to cellular reprogramming [9]. Use of Arid3a null mES lines in an in vitro differentiation system, coupled with mouse tissue histology has provided here an insight into Arid3a function in early development and later in organ growth. We have compared the transcriptional profiles of Arid3a-/- and Arid3a+/+ mESC under conditions that retain pluripotency as well as following differentiation. While loss of Arid3a has been shown to induce reprogramming [8,9], Arid3a-deficient mESC retain the ability to differentiate and form all three germ layers. Thus Arid3a works as part of a larger regulatory network that controls differentiation.
Arid3a expression increases gradually with a majority of the protein localized to the nuclear matrix [9]. This gradual increase of Arid3a during early differentiation suggested a transcriptional function. Accordingly, Arid3a knockout and wildtype mES cells were comparable in self-renewal and maintenance of pluripotency, but during differentiation, Arid3a knockout mES cells turned on gene expression of mesoderm and neuroectoderm lineages faster than controls. However, other lineages, including endoderm and ectoderm, were unperturbed. That Arid3a may regulate genes critical to these lineage-specific pathways was confirmed by microarray findings that undifferentiated Arid3a-/- mES upregulated early differentiation and vascular pathways. The requirement of Arid3a for normal mesoderm differentiation is consistent with the need for Arid3a in hematopoietic stem cell and kidney development, as both are mesoderm-derived tissues. In support of this hypothesis, a recent report found that an Arid3a-/- kidney cell line, KKPS5, spontaneously developed into multicellular nephron-like structures in vitro [25]. Further, these cells engrafted into immunocompromised medaka mesonephros, where they formed mouse nephron structures. Thus, Arid3a may provide a new model system for studying kidney development.
Key regulatory mES cell transcription factors exist in a balanced state; for example, too little or too much of Oct4, Sox2, or Nanog can induce differentiation [26]. Loss of Nanog commits cells to the endoderm lineage, while loss of Oct4 commits cells to the trophoectoderm. Yet, raising Oct4 levels by just 50% commits ES cells to the endoderm and mesoderm lineages [27]. Overexpression of Sox2 by as little as twofold can induce lineage differentiation-but down the endoderm lineage [28]. We have shown that loss of Arid3a induces reprogramming in somatic cells and alters the differentiation pattern of mES cells [8,9,29]. Taking into consideration both these observations and the transcriptional balance required for differentiation, we suggest that Arid3a may act, not as a switch, but as a knob to repress levels of key pluripotency factors in a timely and controlled manner during differentiation. Thus, in part, Arid3a controls the timing of differentiation of the developing embryo. This balanced differentiation, when unbalanced through loss of Arid3a, appears to favor mesoderm differentiation by increasing the rate at which mesoderm-specific genes are expressed However, Arid3a loss proves to be problematic during later differentiation. The majority of Arid3a null mice die in utero due to HSC deficiencies [4]. We observed that the rare survivors that overcome early lethality develop normally to a first approximation. However, we now appreciate that there is a proliferative defect in the null kidney.
Our data, along with the aforementioned accounts, indicate that Arid3a is an important regulator of timely differentiation-a role that extends beyond early differentiation and affects multiple tissue types. Further in-depth studies of conditionally-deleted Arid3a, particularly focused on the kidney, are required to determine fully Arid3a effects in adult organs.
Materials and Methods
Derivation of Arid3a Null and heterozygous embryonic stem cells
To obtain Arid3a-/- mES lines, blastocysts were flushed out of the horns of 3.5 day pregnant Arid3a± females which had been mated with Arid3a± males [30]. Blastocysts were transferred onto STO feeder layers in mES media (DMEM supplemented with 20% FBS, penicillin/streptomycin, nucleosides, non-essential amino acids, and β-mercaptoethanol) and cultured at 37°C in 5% CO2 in humidified air for 6-7 days without media changes. The inner cell masses were identified, treated with trypsin, disrupted, and then transferred individually and subcultured in 24-well STO feeder plates. Four days later, single cell clones of compact mES colonies were passaged onto 6-well plates and then split after 2–3 generations for confirmation of null genotype by PCR.
Embryoid body formation assay
Embryonic stem cell colonies were lightly trypsinized, feeder cells were removed, and cells were resuspended in mES media that contained no LIF at a concentration of 20,000 cells/ml. Approximately 80-20ul droplets (each containing ~400 cells) were placed on the lid of a petri dish. The lid was then inverted and placed over the petri dish filled with PBS to maintain proper humidity. Hanging drops were kept in standard cell culture conditions for three days. On the third day hanging drops were collected into 10ml mES media that contained no LIF on bacterial culture plates to prevent attachment. Media was changed every other day, and EBs were collected by centrifugation at the time points indicated. For adherent experiments, EBs were collected at day 8 and plated onto gelatin coated 10 cm cell culture dishes. Protocol modified from David Stewart lab protocol, personal communication.
Teratoma and tissue histology
Near confluent cells were lightly trypsinized (5% trypsin/1% EDTA) and washed twice in PBS. 100ul containing ~350,000 cells were injected subcutaneously into the flanks of NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice. Mice were palpated for tumor growth and sacrificed when visible tumors were 1-2 cm across the longest diameter. Tissues were harvested from mice at indicated timepoints using humane techniques as defined by the University of Texas at Austin Institutional Animal Care and Use Committee. Tissues were prepared in 5% paraformaldehyde solution for a minimum of 24 hours. The tissues were then dehydrated using 30, 50, 70, 95, and 100% ethanol (EtOH). Samples were stored in 100% EtOH at 4°C until processing. The tissues samples were embedded at the Histology and Tissue Processing Facility Core located at The Virginia Harris Cockrell Cancer Research Center at The University of Texas MD Anderson Cancer Center, Science Park facility. Embedded tissues were sectioned and stained for Ki67 or Hematoxylin and Eosin (H&E) at the core facility. Teratoma samples were analyzed by a trained pathologist at M.D. Anderson – Science Park Histology and Tissue Processing Facility Core.
Microarray analysis
Cells were harvested by trypsin digestion. Total RNA was isolated (Qiagen RNAEasy). On-column DNase digestion was performed (Qiagen) to remove genomic DNA contamination. RNA was reversed transcribed (Invitrogen). Labeling with cy3 random nonamers and array hyArid3adizations were performed by following the Nimblegen expression array protocol. Alignment and data normalization were done using Nimblescan provided from Nimblegen.
Proliferation analysis
Tissues sections stained for Ki67 were imaged using a light microscope under 10x magnification. The entire section was imaged with no overlap. For the spleen, a grid was placed on top of the section image and ten squares were randomly chosen, counted from each picture, and averaged. Whole images of the kidney were counted. The mean from the entire organ was plotted. The variance was determined by F-test; the P-value was determined by T-test; and the standard error of the mean was calculated. All calculations and statistical tests were performed in Microsoft Excel.
Author Contributions
MP, B-KL, VRI and HOT designed research; MP, B-KL, and CR performed research; MP, B-KL VRI, CR, and HOT analyzed data; MP and HOT wrote the manuscript, and VRI and HOT initiated the project.
Acknowledgements
We thank June Harriss for her excellent contribution to all aspects of the animal husbandry, Chhaya Das and Maya Ghosh for help in cell culture, and molecular techniques. We thank members of the Iyer and Tucker laboratories for discussions and reading of the manuscript. Histology was performed at the histology core of the MD Anderson Cancer Center in Smithville, TX. This work was supported in part by grants from the NIH (R01 CA130075) and CPRIT (RP120194) to V.R.I and by NIH Grant R01CA31534, Cancer Prevention Research Institute of Texas (CPRIT) Grants (RP100612, RP120348) and the Marie Betzner Morrow Centennial Endowment to H.O.T.
References
- Martello G, Smith A. The nature of embryonic stem cells. Annu Rev Cell Dev Biol. 2014; 30; 647-675. Ref.: https://goo.gl/2fQ2im
- Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat Rev Mol Cell Biol. 2009; 10; 526-537. Ref.: https://goo.gl/zmVgwx
- Nixon JC, Ferrell S, Miner C, Oldham AL, Hochgeschwender U, et al. Transgenic mice expressing dominant-negative Arid3a exhibit defects in B1 B cells. J Immunol Baltim Md. 2008; 181; 6913-6922. Ref.: https://goo.gl/suWSPu
- Webb CF, Bryant J, Popowski M, Allred L, Kim D, et al. The ARID family transcription factor Arid3a is required for both hematopoietic stem cell and B lineage development. Mol Cell Biol. 2011; 31; 1041-1053. Ref.: https://goo.gl/wGgErP
- Kim PG, Canver MC, Rhee C, Ross SJ, Harriss JV, et al. Interferon-α signaling promotes embryonic HSC maturation. Blood. 2016; 128: 204-216. Ref.: https://goo.gl/nTGHsW
- Rhee C, Edwards M, Dang C, Harris J, Brown M, et al. ARID3A is required for mammalian placenta development. Dev Biol. 2017; 422: 83-91. Ref.: https://goo.gl/Dq2rku
- Rhee C, Lee BK, Beck S, Anjum A, Cook KR, Popowski M, et al. Corrigendum: Arid3a is essential to execution of the first cell fate decision via direct embryonic and extraembryonic transcriptional regulation. Genes Dev. 2015; 29: 1890. Ref.: https://goo.gl/aSTe3c
- An G, Miner CA, Nixon JC, Kincade PW, Bryant J, et al. Loss of Arid3a/ARID3a function promotes developmental plasticity. Stem Cells Dayt Ohio. 2010; 28: 1560-1567. Ref.: https://goo.gl/NFFfUf
- Popowski M, Templeton TD, Lee BK, Rhee C, Li H, et al. Arid3a/Arid3A Acts as a Barrier to Somatic Cell Reprogramming through Direct Regulation of Oct4, Sox2, and Nanog. Stem Cell Rep. 2014; 2: 26-35. Ref.: https://goo.gl/ZUgmtk
- Wang J, Rao S, Chu J, Shen X, Levasseur DN, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006; 444: 364-368. Ref.: https://goo.gl/aWnkHB
- Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, et al. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med. 2000; 6: 88-95. Ref.: https://goo.gl/Vrwepk
- Kurosawa H. Methods for Inducing Embryoid Body Formation: In Vitro Differentiation System of Embryonic Stem Cells. J Biosci Bioeng. 2007; 103: 389-398. Ref.: https://goo.gl/YhYLHo
- Wang R, Clark R, Bautch VL. Embryonic stem cell-derived cystic embryoid bodies form vascular channels: an in vitro model of blood vessel development. Dev Camb Engl. 1992; 114: 303-316. Ref.: https://goo.gl/daxwVF
- Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985; 87: 27-45. Ref.: https://goo.gl/WKVyCQ
- Yasuda E, Seki Y, Higuchi T, Nakashima F, Noda T, et al. Development of cystic embryoid bodies with visceral yolk-sac-like structures from mouse embryonic stem cells using low-adherence 96-well plate. J Biosci Bioeng. 2009; 107: 442-446. Ref.: https://goo.gl/a4Pi8d
- Ng YS, Ramsauer M, Loureiro RMB, D’Amore PA. Identification of genes involved in VEGF-mediated vascular morphogenesis using embryonic stem cell-derived cystic embryoid bodies. Lab Invest. 2004; 84: 1209-1218. Ref.: https://goo.gl/6QALJf
- Fuegemann CJ, Samraj AK, Walsh S, Fleischmann BK, Jovinge S, et al. Differentiation of Mouse Embryonic Stem Cells into Cardiomyocytes via the Hanging-Drop and Mass Culture Methods. Curr Protoc Stem Cell Biol. 2010. Ref.: https://goo.gl/fKxcSo
- Kim D, Tucker PW. A regulated nucleocytoplasmic shuttle contributes to Arid3a’s function as a transcriptional activator of immunoglobulin genes. Mol Cell Biol. 2006; 26: 2187-2201. Ref.: https://goo.gl/JGdVXL
- Schmidt C, Kim D, Ippolito GC, Naqvi HR, Probst L, et al. Signalling of the BCR is regulated by a lipid rafts-localised transcription factor, Bright. EMBO J. 2009; 28: 711-724. Ref.: https://goo.gl/9ehCcC
- Ma K, Araki K, Ichwan SJ, Suganuma T, Tamamori-Adachi M, et al. E2FBP1/DRIL1, an AT-rich interaction domain-family transcription factor, is regulated by p53. Mol. Cancer Res. 2003; 1: 438-444. Ref.: https://goo.gl/c9pZrR
- Peeper DS, Shvarts A, Brummelkamp T, Douma S, Koh EY, et al. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell Biol. 2002; 4:148-153. Ref.: https://goo.gl/8fQDEL
- Lin L, Zhou Z, Zheng L, Alber S, Watkins S, et al. Cross talk between Id1 and its interactive protein Dril1 mediate fibroblast responses to transforming growth factor-beta in pulmonary fibrosis. Am J Pathol. 2008; 173: 337-346. Ref.: https://goo.gl/uCbTzr
- Zong RT, Tucker PW, Das C. Regulation of matrix attachment region-dependent, lymphocyte-restricted transcription through differential localization within promyelocytic leukemia nuclear bodies. EMBO J. 2000; 19: 4123-4133. Ref.: https://goo.gl/W31L98
- Lin D, Ippolito GC, Zong RT, Bryant J, Koslovsky J, et al. Arid3a/ARID3A contributes to chromatin accessibility of the immunoglobulin heavy chain enhancer. Mol Cancer. 2007; 6: 23. Ref.: https://goo.gl/jAvdqX
- Webb CF, Ratliff ML, Powell R, Wirsig-Wiechmann CR, Lakiza O, et al. A developmentally plastic adult mouse kidney cell line spontaneously generates multiple adult kidney structures. Biochem Biophys Res Commun. 2015; 463: 1334-1340. Ref.: https://goo.gl/o3Qa7N
- Bosnali M, Münst B, Thier M, Edenhofer F. Deciphering the stem cell machinery as a basis for understanding the molecular mechanism underlying reprogramming. Cell Mol Life Sci. 2009; 66: 3403-3420. Ref.: https://goo.gl/FTXrNa
- Chambers I. The Molecular Basis of Pluripotency in Mouse Embryonic Stem Cells. Cloning Stem Cells. 2004; 6, 386-391. Ref.: https://goo.gl/6YBo8d
- Kopp JL, Ormsbee BD, Desler M, Rizzino A. Small Increases in the Level of Sox2 Trigger the Differentiation of Mouse Embryonic Stem Cells. STEM CELLS. 2008; 26: 903-911. Ref.: https://goo.gl/zKL2TW
- Rhee C, Lee BK, Beck S, Anjum A, Cook KR, et al. Arid3a is essential to execution of the first cell fate decision via direct embryonic and extraembryonic transcriptional regulation. Genes Dev. 2014; 28: 2219-2232. Ref.: https://goo.gl/GV6Bb3
- Nagy A. Manipulating the Mouse Embryo: A Laboratory Manual, Third Edition. 2002. Ref.: https://goo.gl/vpdDo1